

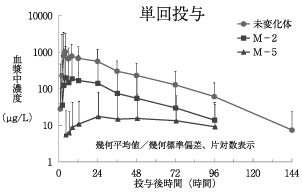
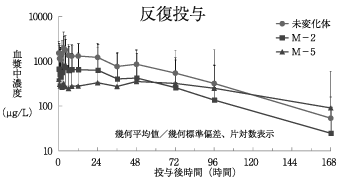
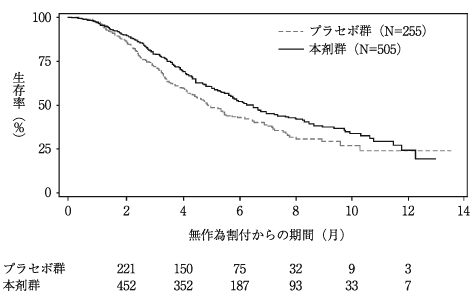
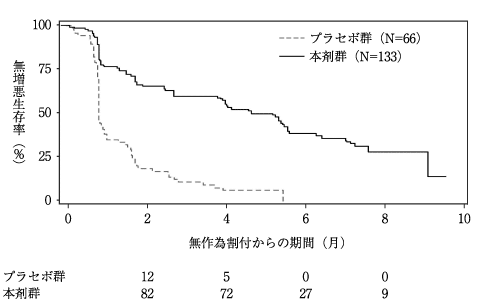
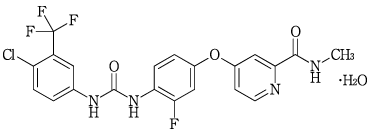
1
社内資料: ウサギにおける胚・胎児発生に関する毒性試験(2013年3月25日承認、CTD2.6.6.6.3)
2
社内資料: ラットにおける乳汁中分泌に関する試験(2013年3月25日承認、CTD2.6.4.6.4)
3
NDBを用いた調査結果の概要(VEGF/VEGFR阻害作用を有する薬剤の動脈解離に関するリスク評価): https://www.pmda.go.jp/files/000266521.pdf
4
社内資料: 日本人進行固形がん患者を対象とした国内第Ⅰ相臨床試験(2013年3月25日承認、CTD2.7.6.6)
5
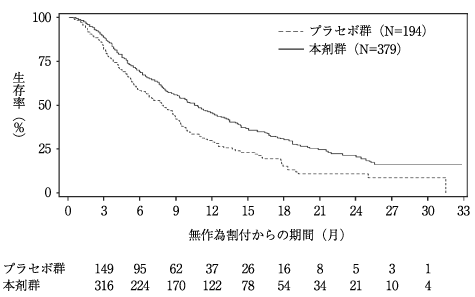
社内資料: 転移性結腸・直腸癌患者を対象とした国際共同第Ⅲ相臨床試験(2013年3月25日承認、CTD2.7.6.12)
6
社内資料: 消化管間質腫瘍患者を対象とした国際共同第Ⅲ相臨床試験(2013年8月20日承認、CTD2.7.6.1)
7
社内資料: 切除不能な肝細胞癌患者を対象とした国際共同第Ⅲ相臨床試験(2017年6月26日承認、CTD2.7.6.4)
8
Wilhelm SM, et al.: Int J Cancer. 2011; 129: 245-255
9
社内資料: キナーゼ阻害に関する試験(2013年3月25日承認、CTD2.6.2.2.1.4)
10
George S, et al.: J Clin Oncol. 2012; 30: 2401-2407
11
社内資料: ヒト結腸癌異種移植モデルにおけるレゴラフェニブ及び代謝物の作用に関する試験(2013年3月25日承認、CTD2.6.2.2.2.6.1)
12
社内資料: 消化管間質腫瘍モデルに対する作用に関する試験(2013年8月20日承認、CTD2.6.2.2.2.1)
13
社内資料: マウス肝細胞癌同所性移植モデルに対する作用に関する試験(2013年3月25日承認、CTD2.6.2.2.2.3)

