

1
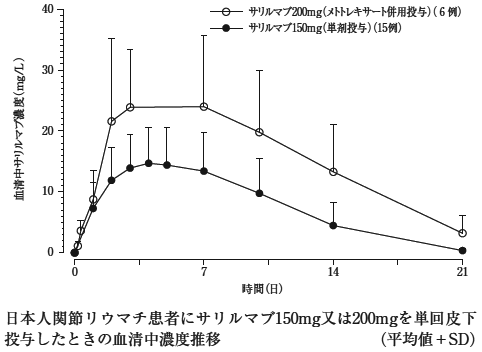
社内資料:国内第1相単回投与試験(メトトレキサート併用試験)(2017年9月27日承認、CTD2.7.2.2)
2
社内資料:国内第1相単回投与試験(単剤投与試験)(2017年9月27日承認、CTD2.7.2.2)
3
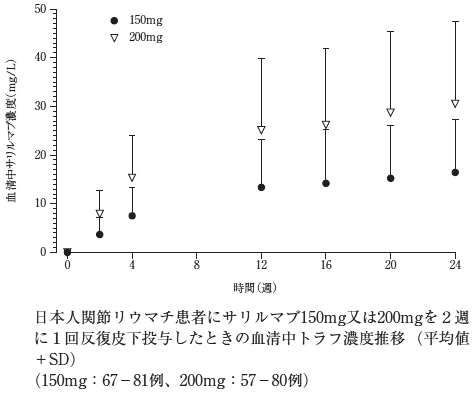
社内資料:国内第2/3相プラセボ対照二重盲検比較試験(メトトレキサート併用試験)(2017年9月27日承認、CTD2.7.2.2)
4
社内資料:海外第1相薬物相互作用試験(シンバスタチン)(2017年9月27日承認、CTD2.7.2.2)
5
社内資料:国内第2/3相プラセボ対照二重盲検比較試験(メトトレキサート併用試験)(2017年9月27日承認、CTD2.7.3.2)
6
社内資料:国内第2/3相プラセボ対照二重盲検比較試験(メトトレキサート併用試験)(2017年9月27日承認、CTD2.7.6.2)
7
社内資料:国内第3相二重盲検試験(メトトレキサート以外のDMARDs併用又は単剤投与試験)(2017年9月27日承認、CTD2.7.3.2)
8
社内資料:国内第3相二重盲検試験(メトトレキサート以外のDMARDs併用又は単剤投与試験)(2017年9月27日承認、CTD2.7.6.2)
9
社内資料:海外第3相プラセボ対照二重盲検比較試験(メトトレキサート併用試験)(2017年9月27日承認、CTD2.7.3.2)
10
社内資料:海外第3相プラセボ対照二重盲検比較試験(メトトレキサート併用試験)(2017年9月27日承認、CTD2.7.6.2)

