1
van Dyck C. H. et al.:N. Engl. J. Med., 2023;388(1):9-21[LEQ-0002]
2
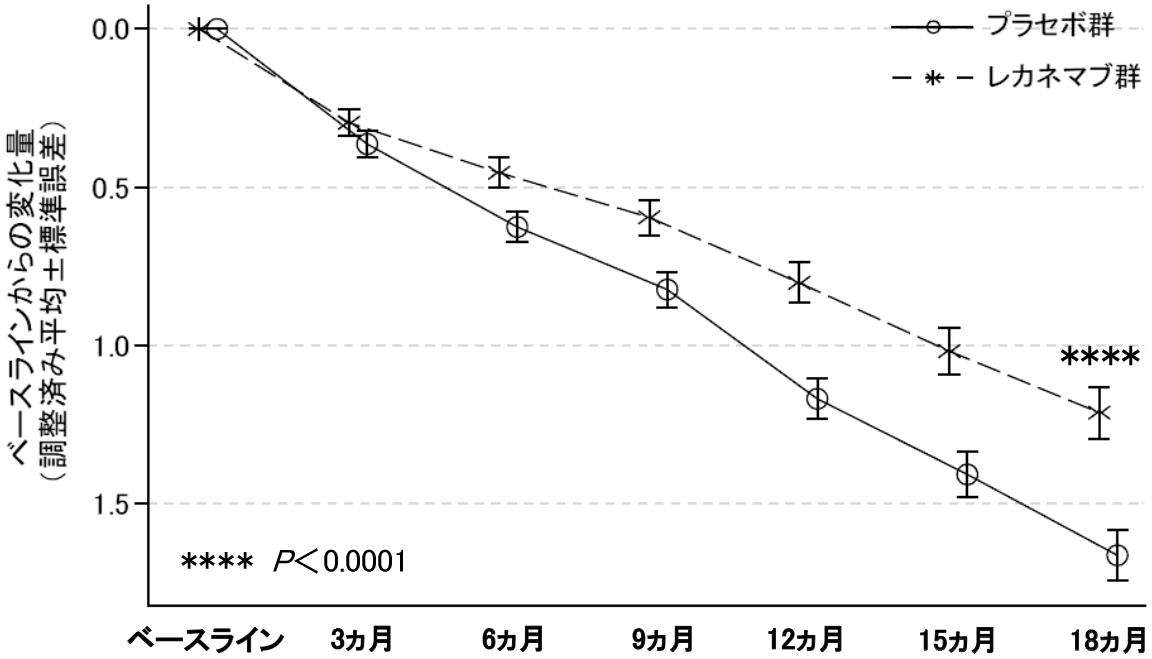
社内資料:国際共同臨床第III相試験(301試験)(2023年9月25日承認、CTD 2.7.6.5)[LEQ-0009]
3
Nicholas J. R. et al.:N. Engl. J. Med., 2023;388(5):478-479[LEQ-0031]
4
社内資料:国内104試験(2023年9月25日承認、CTD 2.7.2.2.2.1.2)[LEQ-0010]
5
社内資料:母集団薬物動態解析(2023年9月25日承認、CTD 2.7.2.3.2)[LEQ-0015]
6
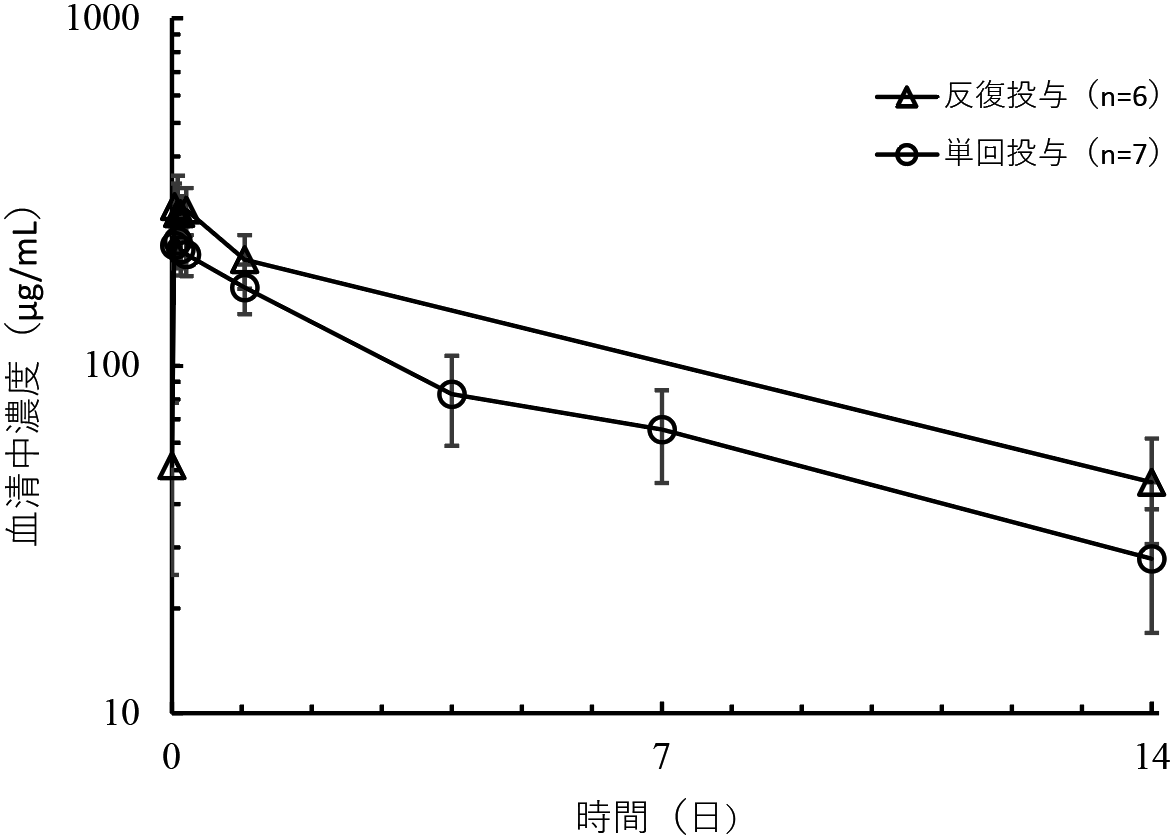
社内資料:薬物動態プロファイル(2023年9月25日承認、CTD 2.7.2.3.1)[LEQ-0016]
7
Swanson C. J. et. al.:Alz. Res. Therapy, 2021;13, 80[LEQ-0001]
8
社内資料:国際共同臨床第II相試験(201試験)(2023年9月25日承認、CTD 2.7.6.3)[LEQ-0011]
9
社内資料:種々アミロイドβへの結合性比較(2023年9月25日承認、CTD 2.6.2.2.1.1)[LEQ-0017]
10
社内資料:ラット海馬神経細胞へのアミロイドβ凝集体の結合阻害(2023年9月25日承認、CTD 2.6.2.2.1.8)[LEQ-0018]
11
社内資料:ミクログリアによるアミロイドβ除去に対する効果(2023年9月25日承認、CTD 2.6.2.2.1.15)[LEQ-0030]
12
社内資料:APPNL-G-Fマウスにおける脳内アミロイドβに対する効果(2023年9月25日承認、CTD 2.6.2.2.2.3)[LEQ-0019]