

1
社内資料: 薬物動態(単回硝子体内投与、外国人)(2012年9月28日承認、CTD2.7.6.1)
2
社内資料: 薬物動態(未熟児網膜症患者)(2022年9月26日承認、CTD2.7.2)
3
社内資料: 薬物動態(第Ⅲ相国際共同試験)(2012年9月28日承認、CTD2.7.2.2.4.5)
4
社内資料: 薬物動態(ウサギ単回硝子体内投与)(2012年9月28日承認、CTD2.6.4.4.2)
5
社内資料: 第Ⅲ相試験(視力に関する評価、併合解析)(2012年9月28日承認、CTD2.7.3.3.2.1)
6
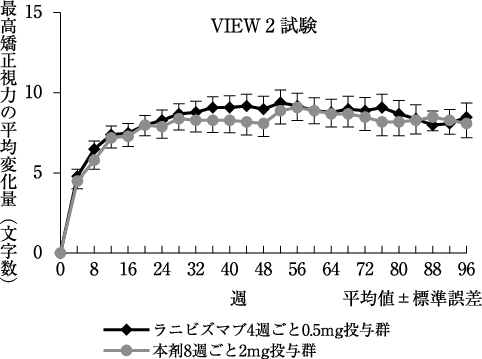
社内資料: 日本人を含む第Ⅲ相国際共同試験(VIEW2試験、1年目)(2012年9月28日承認、CTD2.7.6.12)
7
社内資料: 日本人を含む第Ⅲ相国際共同試験(VIEW2試験、2年目)(2012年9月28日承認、CTD2.7.6.19)
8
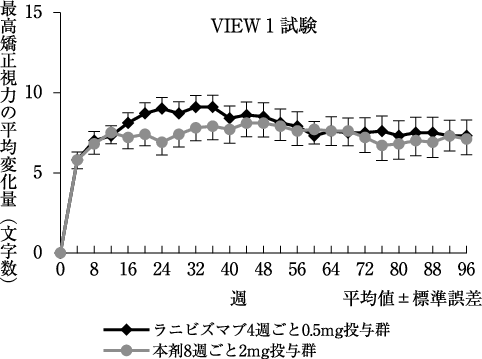
社内資料: 海外第Ⅲ相試験(VIEW1試験、1年目)(2012年9月28日承認、CTD2.7.6.11)
9
社内資料: 海外第Ⅲ相試験(VIEW1試験、2年目)(2012年9月28日承認、CTD2.7.6.18)
10
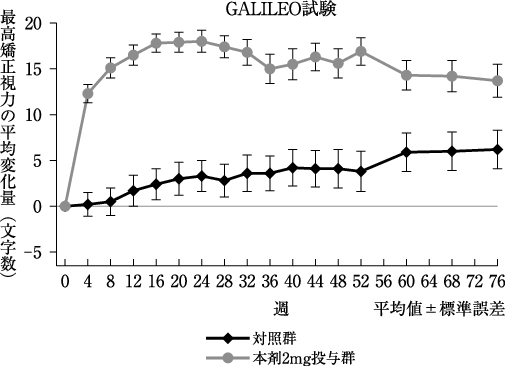
社内資料: 日本人を含む第Ⅲ相国際共同試験(GALILEO試験)(2013年11月22日承認、CTD2.7.6.2)
11
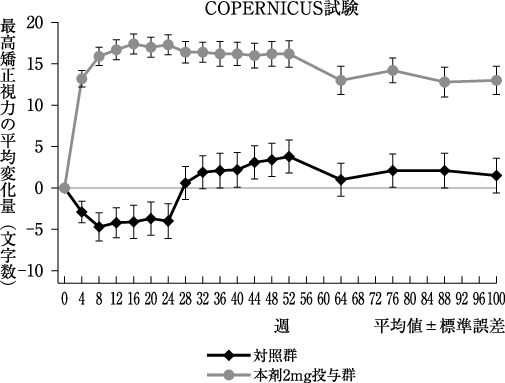
社内資料: 海外第Ⅲ相試験(COPERNICUS試験)(2013年11月22日承認、CTD2.7.6.1)
12
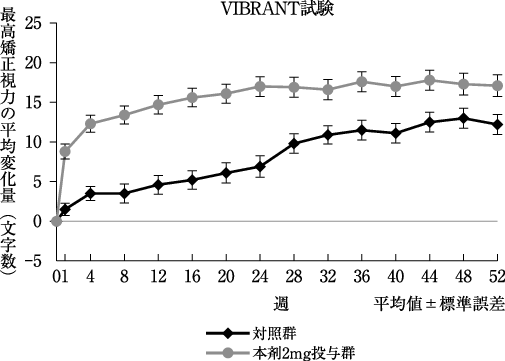
社内資料: 日本人を含む第Ⅲ相国際共同試験(VIBRANT試験)
13
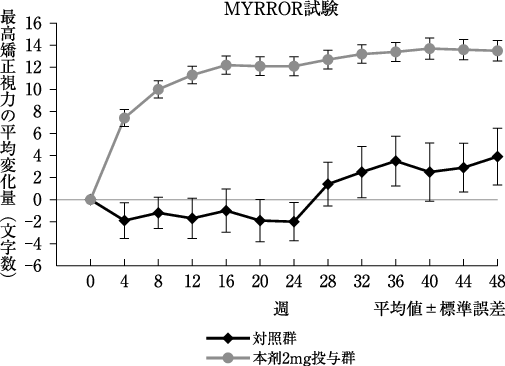
社内資料: 日本人を含む第Ⅲ相国際共同試験(MYRROR試験)(2014年9月19日承認、CTD2.7.6.1)
14
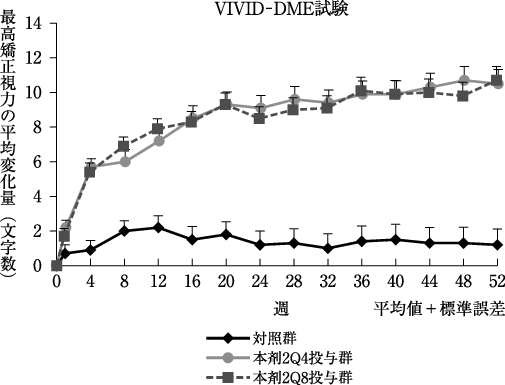
社内資料: 日本人を含む第Ⅲ相国際共同試験(VIVID-DME試験)(2014年11月18日承認、CTD2.7.6.5)
15
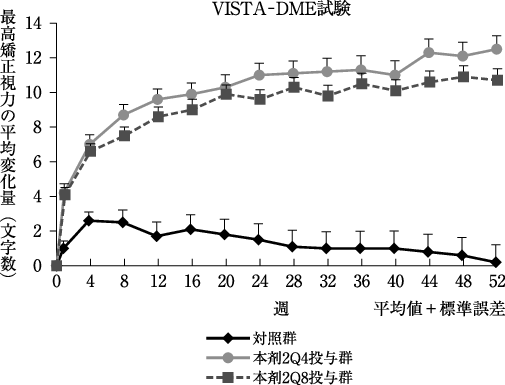
社内資料: 海外第Ⅲ相試験(VISTA-DME試験)(2014年11月18日承認、CTD2.7.6.4)
16
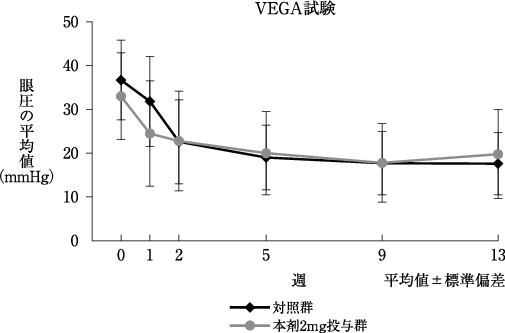
社内資料: 国内第Ⅲ相試験(VEGA試験)(2020年3月25日承認、CTD2.7.6.1)
17
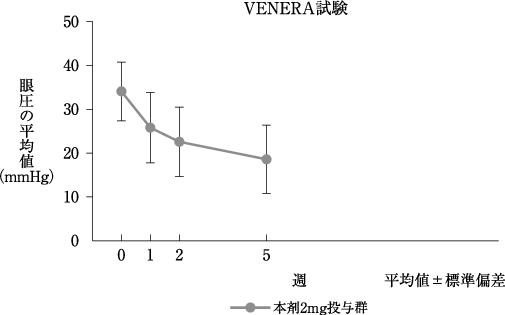
社内資料: 国内第Ⅲ相試験(VENERA試験)(2020年3月25日承認、CTD2.7.6.2)
18
社内資料: 日本人を含む第Ⅲ相国際共同試験(FIREFLEYE試験)(2022年9月26日承認、CTD2.7.6.1)
19
Stahl A, et al.: JAMA. 2022; 328: 348-359
20
Luttun A, et al.: Biochem Biophys Res Commun. 2002; 295: 428-434
21
Cao Y: Sci Signal. 2009; 2: re1
22
Zhong X, et al.: Mol Vis. 2011; 17: 492-507
23
社内資料: In vitroにおける作用(2012年9月28日承認、CTD2.6.2.2.1)
24
Cursiefen C, et al.: Invest Ophthalmol Vis Sci. 2004; 45: 2666-2673
25
Cao J, et al.: Invest Ophthalmol Vis Sci. 2010; 51: 6009-6017
26
Nork TM, et al.: Arch Ophthalmol. 2011; 129: 1042-1052

