


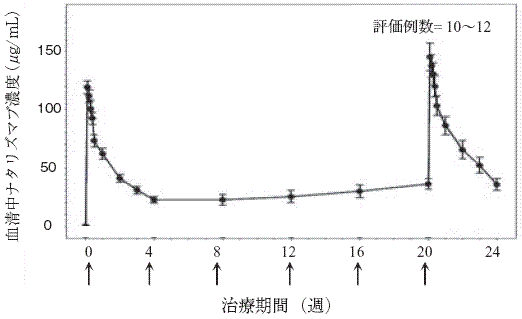
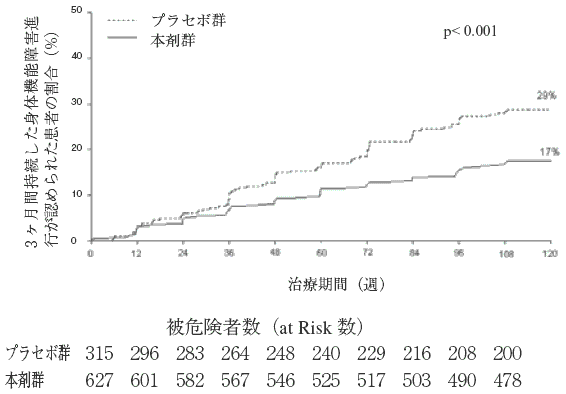
1
タイサブリ®点滴静注300mg適正使用ガイド
2
社内資料:国内第II相臨床試験(2014年3月24日承認、CTD2.7.6.5.1.4)
3
Polman CH et al., N Engl J Med 2006; 354:899-910
4
Rudick RA et al., Expert Rev Neurother 2004; 4:571-80
5
Kent SJ et al., J Neuroimmunol 1995; 58:1-10
6
Kent SJ et al., J Magn Reson Imaging 1995; 5:535-40
7
社内資料:モルモットEAEモデルにおける有効性(2014年3月24日承認、CTD2.6.2.2.8)

