

1
社内資料:第Ⅲ相試験CDI再発率(2017年9月27日承認、CTD 2.5.4.3、CTD 2.7.6.3)
2
社内資料:トキシンBとの結合(2017年9月27日承認、CTD 2.6.2.2)
3
社内資料:臨床分離株での抗トキシン作用(2017年9月27日承認、CTD 2.6.2.2)

抗Clostridium difficileトキシンBヒトモノクローナル抗体
1瓶 335839円
有効成分 | (1バイアル中) ベズロトクスマブ(遺伝子組換え)注1)注2) 625mg/25mL |
|---|---|
添加剤 | 塩化ナトリウム(220mg)、クエン酸ナトリウム水和物(120mg)、クエン酸水和物(20mg)、ポリソルベート80(6.3mg)、ジエチレントリアミン五酢酸(0.20mg)、水酸化ナトリウム(適量) |
| 剤形 | 注射剤(バイアル) |
|---|---|
| pH | 5.7~6.3 |
| 浸透圧比 | 約1(生理食塩液対比) |
| 性状 | 無色~うすい黄色で澄明~うすい乳白色の液 |
0.2~2%未満 | |
|---|---|
神経系障害 | 頭痛、味覚異常、錯感覚 |
血管障害 | 潮紅、ほてり、高血圧 |
胃腸障害 | 悪心、下痢 |
全身障害及び投与局所様態 | 疲労、発熱、注入部位そう痒感 |
臨床検査 | ALT増加、AST増加 |
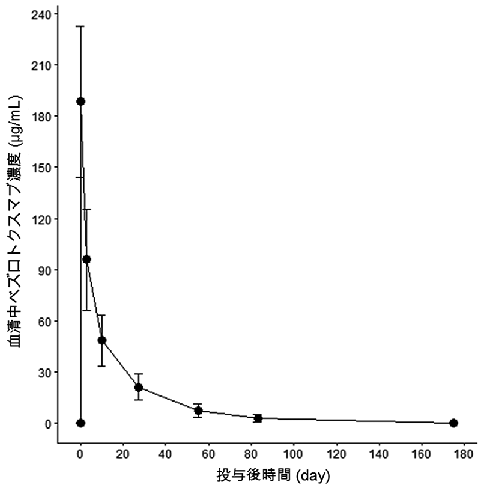
例数 | AUC0-∞ (μg・h/mL) | Cmax (μg/mL)注) | t1/2 (day) | CL (L/day) | Vd (L) |
24 | 52000 (30.5) | 169 (34.2) | 18.2 (26.0) | 0.248 (28.6) | 6.52 (22.9) |
001試験 | 002試験 | |||
本剤群 | プラセボ群 | 本剤群 | プラセボ群 | |
CDI再発率注1) | 17.4% (67/386例) | 27.6% (109/395例) | 15.7% (62/395例) | 25.7% (97/378例) |
プラセボ群との群間差 [95%CI]注2) | -10.1 [-15.9,-4.3] | -9.9 [-15.5,-4.3] | ||
片側P値注2),注3) | 0.0003 | 0.0003 | ||
本剤群 | プラセボ群 | プラセボ群との群間差 (95%CI) | ||
リスク因子の有無 | あり | 16.9% (100/592例) | 29.8% (174/583例) | -13.0 (-17.7,-8.2) |
なし | 15.3% (29/189例) | 16.8% (32/190例) | -1.5 (-9.0,6.0) | |
年齢 | 65歳以上 | 15.4% (60/390例) | 31.4% (127/405例) | -16.0 (-21.7,-10.2) |
65歳未満 | 17.6% (69/391例) | 21.5% (79/368例) | -3.8 (-9.5,1.8) | |
過去6ヵ月間のCDI既往歴 | あり | 25.0% (54/216例) | 41.1% (90/219例) | -16.1 (-24.7,-7.3) |
なし | 13.5% (75/556例) | 20.9% (114/545例) | -7.4 (-11.9,-3.0) | |
免疫不全状態 | あり | 14.6% (26/178例) | 27.5% (42/153例) | -12.8 (-21.7,-4.1) |
なし | 17.1% (103/603例) | 26.5% (164/620例) | -9.4 (-14.0,-4.8) | |
CDI重症度注4) | 重症 | 10.7% (13/122例) | 22.4% (28/125例) | -11.7 (-21.1,-2.5) |
非重症 | 17.5% (110/629例) | 27.6% (169/613例) | -10.1 (-14.7,-5.5) | |
強毒株(リボタイプ027、078又は244)への感染 | あり | 21.6% (22/102例) | 32.2% (37/115例) | -10.6 (-22.1,1.3) |
なし | 16.7% (64/384例) | 29.5% (109/369例) | -12.9 (-18.8,-6.9) | |
リボタイプ027への感染 | あり | 23.6% (21/89例) | 34.0% (34/100例) | -10.4 (-23.0,2.6) |
なし | 16.4% (65/397例) | 29.2% (112/384例) | -12.8 (-18.6,-7.0) | |

