





1
社内報告書:ラット・胚・胎児発生試験(2021年9月27日承認 CTD 2.6.6.6.1)(DIR200240)
2
社内報告書:代謝酵素・薬物動態試験(2021年9月27日承認 CTD 2.7.2.2.1.4)(DIR200241)
3
社内報告書:ラット・3カ月投与毒性試験(2021年9月27日承認 CTD 2.6.6.3.1.3)(DIR200239)
4
社内報告書:遺伝毒性試験(2021年9月27日承認 CTD 2.6.6.4)(DIR210016)
5
社内報告書:局所進行性又は転移性尿路上皮癌患者・国内第Ⅰ相試験(EV-102 study)(2021年9月27日承認 CTD 2.7.6.3)(DIR200242)
6
社内報告書:血漿蛋白結合・薬物動態試験(2021年9月27日承認 CTD 2.7.2.2.1.1)(DIR200243)
7
社内報告書:ラット・排泄・薬物動態試験(2021年9月27日承認 CTD 2.6.4.6.1)(DIR210048)
8
社内報告書:患者・生理学的薬物動態モデル解析(2021年9月27日承認 CTD 2.7.2.2.5.1)(DIR200246)
9
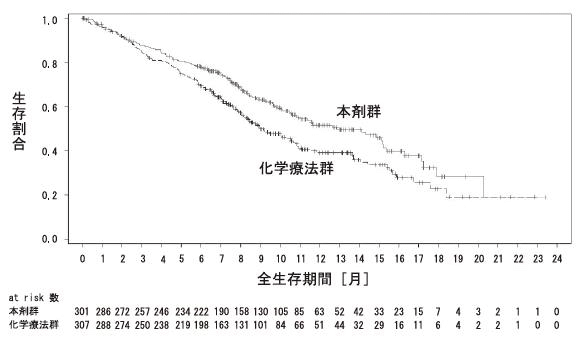
社内報告書:局所進行性又は転移性尿路上皮癌患者・国際共同第Ⅲ相試験(EV-301 study)(2021年9月27日承認 CTD 2.7.6.4)(DIR200247)
10
社内報告書:癌細胞(ヒト由来)・薬理作用(ヒトNectin-4への結合親和性)(2021年9月27日承認 CTD 2.6.2.2.3.1)(DIR200249)
11
社内報告書:膀胱癌細胞(ヒト由来)・薬理作用(膀胱癌における細胞内移行と輸送)(2021年9月27日承認 CTD 2.6.2.2.3.2)(DIR200250)
12
社内報告書:膀胱癌細胞(ヒト由来)・薬理作用(MMAEの細胞内放出)(2021年9月27日承認 CTD 2.6.2.2.3.3)(DIR200251)
13
Francisco JA, et al.:Blood. 2003;102(4):1458-1465[R–08448]
14
社内報告書:癌細胞(ヒト由来)・薬理作用(細胞傷害活性)(2021年9月27日承認 CTD 2.6.2.2.4.1)(DIR200252)
15
社内報告書:膀胱癌細胞(ヒト由来)・薬理作用(2次的バイスタンダー効果)(2021年9月27日承認 CTD 2.6.2.2.4.4)(DIR200253)
16
社内報告書:膀胱癌患者由来腫瘍移植SCIDマウス・薬理作用(2021年9月27日承認 CTD 2.6.2.2.5.1)(DIR200254)

