


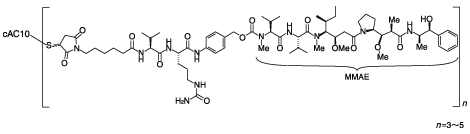
1
ブレンツキシマブ ベドチンの反復投与毒性試験(2014年1月17日承認、CTD 2.6.6.3)
2
ブレンツキシマブ ベドチンの生殖発生毒性試験(2014年1月17日承認、CTD 2.6.6.6)
3
Duggan DB et al.: J Clin Oncol.2003;21(4):607-614.
4
Martin WG et al.: J Clin Oncol.2005;23(30):7614-7620.
5
Hoskin PJ et al.: J Clin Oncol.2009;27(32):5390-5396.
6
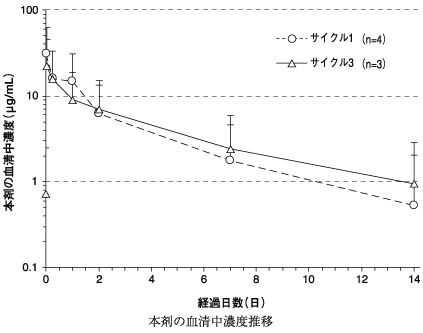
ブレンツキシマブ ベドチンの臨床薬理試験成績(2014年1月17日承認、CTD 2.7.2.2、2.7.2.3)
7
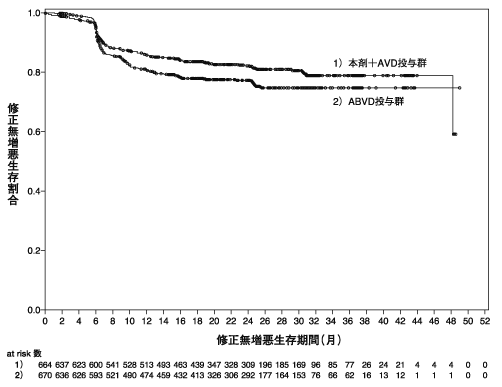
ブレンツキシマブ ベドチンの国際共同第Ⅲ相試験成績①(社内資料)
8
ブレンツキシマブ ベドチンの国内第Ⅰ/Ⅱ相試験成績(2014年1月17日承認、CTD 2.7.2.2、2.7.6.5)
9
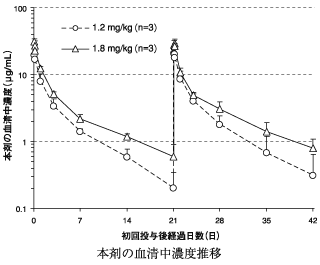
ブレンツキシマブ ベドチンの非臨床薬物動態試験成績(2014年1月17日承認、CTD 2.6.4.4、2.6.4.5)
10
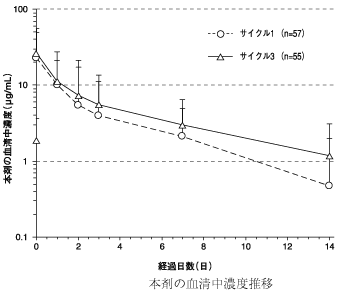
ブレンツキシマブ ベドチンの国際共同第Ⅰ/Ⅱ相試験成績(2022年5月26日承認、CTD 2.7.2.2、2.7.6.1)
11
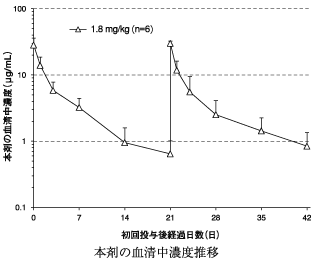
ブレンツキシマブ ベドチンの国内第Ⅰ相試験成績(社内資料)
12
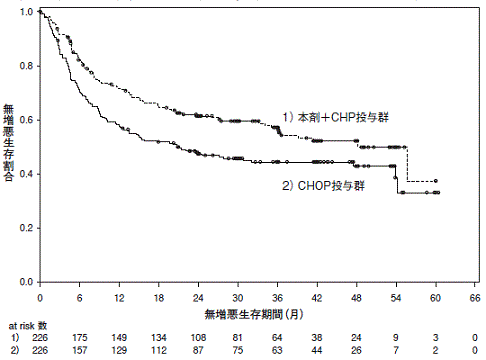
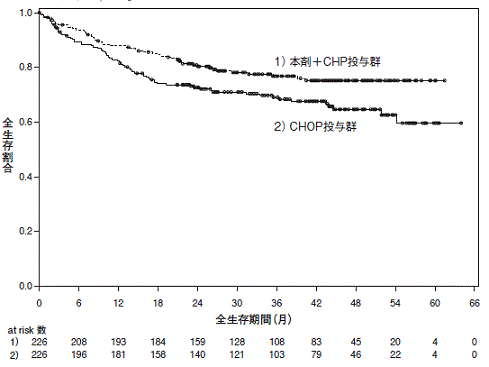
ブレンツキシマブ ベドチンの国際共同第Ⅲ相試験成績②(社内資料)
13
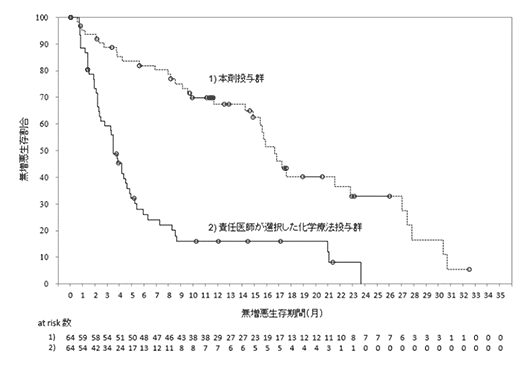
ブレンツキシマブ ベドチンの海外臨床試験成績①(2014年1月17日承認、CTD 2.7.3.2、2.7.6.3)
14
ブレンツキシマブ ベドチンの海外臨床試験成績②(2014年1月17日承認、CTD 2.7.6.4)
15
ブレンツキシマブ ベドチンの海外臨床試験成績③(社内資料)
16
ブレンツキシマブ ベドチンの海外臨床試験成績④(社内資料)
17
ブレンツキシマブ ベドチンの国内第II相試験成績(2023年11月24日承認、CTD 2.7.6.2)
18
ブレンツキシマブ ベドチンの海外臨床試験成績⑤(2023年11月24日承認、CTD 2.7.6.1)
19
Katz J et al.: Clin Cancer Res., 17(20):6428-6436, 2011.
20
ブレンツキシマブ ベドチンの薬効薬理試験成績(2014年1月17日承認、CTD 2.6.2.2)
21
ブレンツキシマブ ベドチンの非臨床薬理試験成績(2014年1月17日承認、CTD 2.6.2.2)

