


1
日本小児腎臓病学会編 小児特発性ネフローゼ症候群診療ガイドライン2020
2
医療上の必要性の高い未承認薬・適応外薬検討会議 公知申請への該当性に係る報告書:リツキシマブ(遺伝子組換え)(免疫抑制状態下のCD20陽性のB細胞性リンパ増殖性疾患(成人))
3
医療上の必要性の高い未承認薬・適応外薬検討会議 公知申請への該当性に係る報告書:リツキシマブ(遺伝子組換え)(免疫抑制状態下のCD20陽性のB細胞性リンパ増殖性疾患(小児))
4
Krysko KM, et al.:Neurol Neuroimmunol Neuroinflamm. 2020;7(1):e637
5
Igarashi T, et al.:Ann. Oncol. 2002;13:928-943
6
「IDEC-C2B8 CD20陽性のB細胞性非ホジキンリンパ腫国内臨床試験及び海外臨床試験の概要」(社内資料)
7
Igarashi T, et al.:Int. J. Hematol. 2001;73:213-221
8
Tobinai K, et al.:Ann. Oncol. 1998;9:527-534
9
米国添付文書 2001
10
「IDEC-C2B8 CD20陽性の慢性リンパ性白血病国内臨床試験及び海外臨床試験の概要」(承認年月日:2019年3月26日、CTD 2.5.3.3.2)
11
「IDEC-C2B8難治性のネフローゼ症候群(頻回再発型あるいはステロイド依存性を示す場合)国内臨床試験の概要」(承認年月日:2014年8月29日、CTD 2.5.3.3.2)
12
「IDEC-C2B8全身性強皮症国内臨床試験の概要」(承認年月日:2021年9月27日、CTD 2.7.2.2.3)
13
「IDEC-C2B8難治性天疱瘡(腫瘍随伴性天疱瘡を除く)国内臨床試験の概要」(承認年月日:2021年12月24日、CTD 2.5.3.3.2)
14
「IDEC-C2B8視神経脊髄炎スペクトラム障害の再発予防国内臨床試験の概要」(承認年月日:2022年6月20日、CTD 2.5.3.3.3)
15
「IDEC-C2B8 ABO血液型不適合腎移植国内臨床試験の概要」(承認年月日:2016年2月29日、CTD 2.5.3.3.2)
16
「抗ドナー抗体陽性、抗HLA抗体陽性の生体腎移植の抗体関連型拒絶反応抑制国内臨床試験の概要」(CTD 2.5.3.3.2)
17
Alasfoor K, et al.:Ann. Hematol. 2009;88:239-243
18
「CD20陽性のB細胞性非ホジキンリンパ腫におけるIDEC-C2B8薬物動態」(社内資料)
19
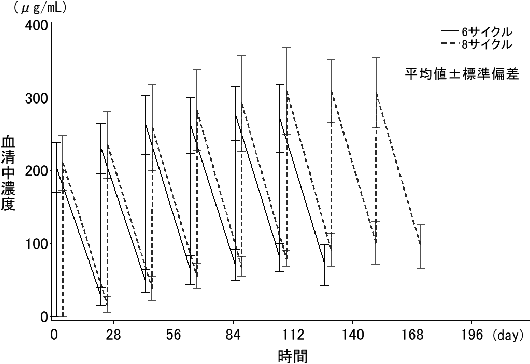
「CD20陽性の慢性リンパ性白血病におけるIDEC-C2B8薬物動態」(承認年月日:2019年3月26日、CTD 2.7.2.2)
20
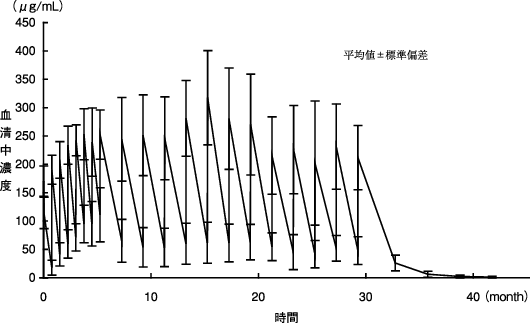
「IDEC-C2B8難治性のネフローゼ症候群(頻回再発型あるいはステロイド依存性を示す場合)国内臨床試験の概要」(承認年月日:2014年8月29日、CTD 2.7.2.2.2.2.1)
21
「IDEC-C2B8全身性強皮症国内臨床試験の概要」(承認年月日:2021年9月27日、CTD 2.7.2.2.1)
22
「IDEC-C2B8難治性天疱瘡(腫瘍随伴性天疱瘡を除く)国内臨床試験の概要」(承認年月日:2021年12月24日、CTD 2.7.2.2.2)
23
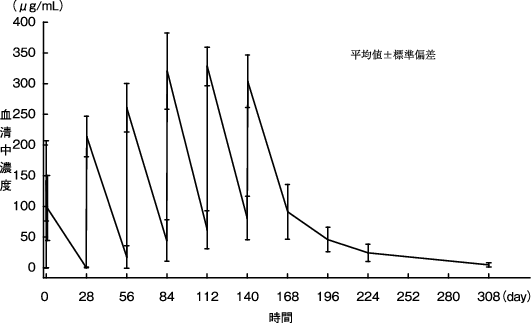
「ABO血液型不適合腎移植におけるIDEC-C2B8薬物動態」(承認年月日:2016年2月29日、CTD 2.5.3.2)
24
「抗ドナー抗体陽性、抗HLA抗体陽性の生体腎移植の抗体関連型拒絶反応抑制におけるIDEC-C2B8薬物動態」(CTD 2.7.2.2.1)
25
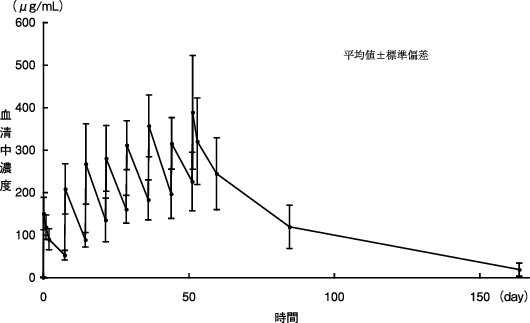
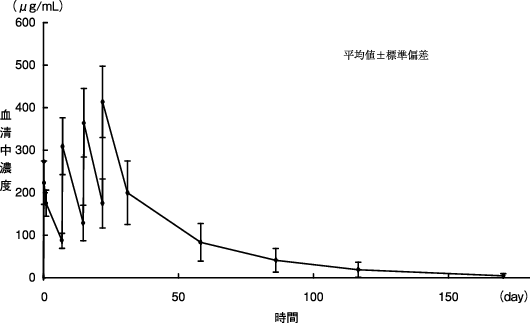
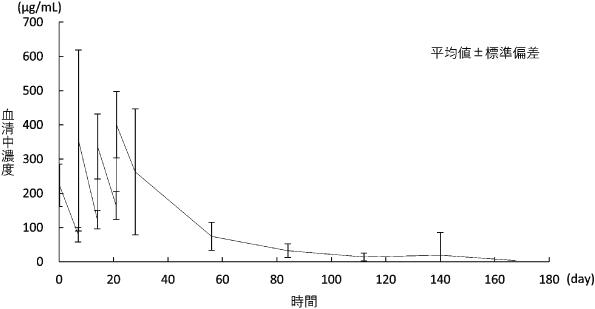
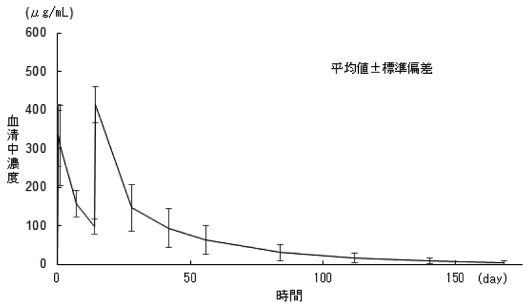
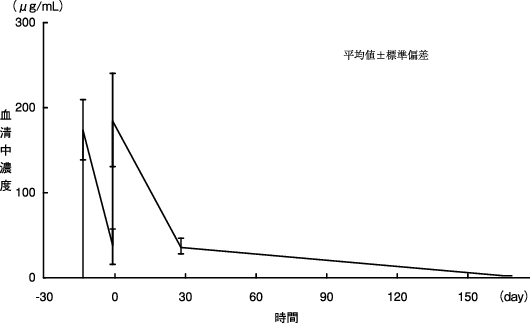
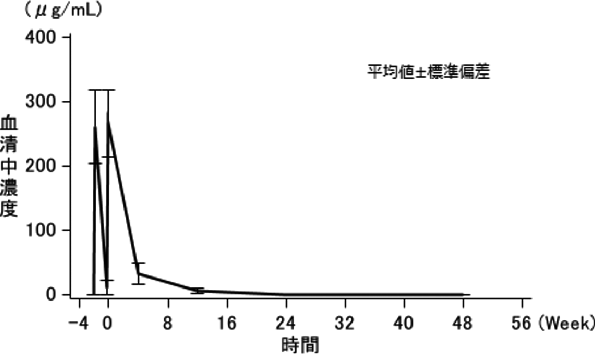
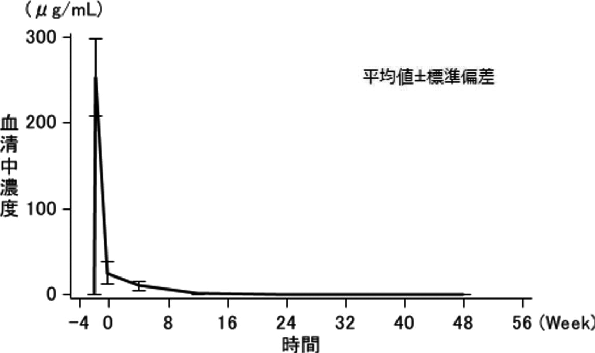
「IDEC-C2B8単回投与時の薬物動態と腫瘍移行性」(承認年月日:2001年6月20日、申請資料概要 ヘ. 吸収、分布、代謝、排泄)
26
「IDEC-C2B8未治療CD20陽性のろ胞性非ホジキンリンパ腫に対する維持療法の海外臨床試験の概要」(社内資料)
27
「IDEC-C2B8再発又は難治性のろ胞性非ホジキンリンパ腫に対する他の抗悪性腫瘍剤との併用の海外臨床試験の概要」(社内資料)
28
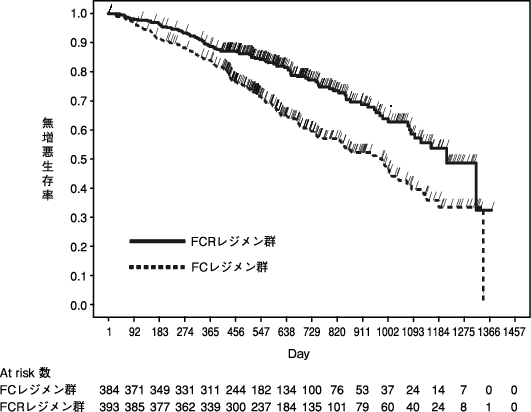
「IDEC-C2B8 CD20陽性の慢性リンパ性白血病国内臨床試験及び海外臨床試験の概要」(承認年月日:2019年3月26日、CTD 2.5.4.3)
29
「IDEC-C2B8未治療CD20陽性の慢性リンパ性白血病国内臨床試験の概要」(社内資料)
30
「IDEC-C2B8未治療CD20陽性の慢性リンパ性白血病海外臨床試験の概要」(社内資料)
31
「IDEC-C2B8再発又は難治性のCD20陽性の慢性リンパ性白血病海外臨床試験の概要」(社内資料)
32
Stone J. et al.:N. Engl. J. Med. 2010;363:221-232
33
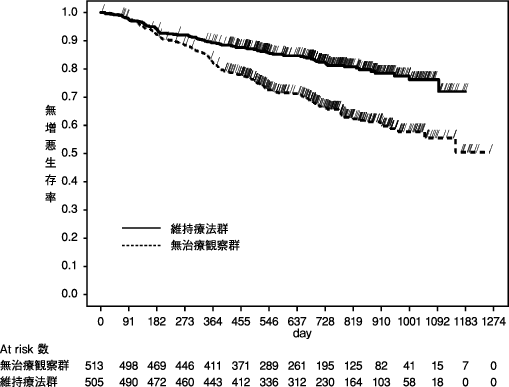
「IDEC-C2B8難治性のネフローゼ症候群(頻回再発型あるいはステロイド依存性を示す場合)国内臨床試験の概要」(承認年月日:2014年8月29日、CTD 2.5.4.3.4)
34
「IDEC-C2B8難治性のネフローゼ症候群(頻回再発型あるいはステロイド依存性を示す場合)国内臨床試験(RCRNS-01試験:中間解析)の概要」(社内資料)
35
「IDEC-C2B8難治性のネフローゼ症候群(頻回再発型あるいはステロイド依存性を示す場合)国内臨床試験の概要」(承認年月日:2014年8月29日、CTD 2.7.4.2.1)
36
「IDEC-C2B8全身性強皮症国内臨床試験の概要」(社内資料)
37
「IDEC-C2B8全身性強皮症国内臨床試験の概要」(承認年月日:2021年9月27日、CTD 2.5.5.2.2)
38
「IDEC-C2B8全身性強皮症国内臨床試験の概要」(承認年月日:2021年9月27日、CTD 2.5.5.3.1)
39
「IDEC-C2B8難治性天疱瘡(腫瘍随伴性天疱瘡を除く)国内臨床試験の概要」(承認年月日:2021年12月24日、CTD 2.7.3.3.2.2)
40
「IDEC-C2B8難治性天疱瘡(腫瘍随伴性天疱瘡を除く)国内臨床試験の概要」(承認年月日:2021年12月24日、CTD 2.7.4.2.1.1)
41
「IDEC-C2B8尋常性天疱瘡海外臨床試験の概要」(承認年月日:2021年12月24日、CTD 2.7.3.3.2.1)
42
「IDEC-C2B8尋常性天疱瘡海外臨床試験の概要」(承認年月日:2021年12月24日、CTD 2.7.4.2.1.1)
43
「IDEC-C2B8視神経脊髄炎スペクトラム障害の再発予防国内臨床試験の概要」(承認年月日:2022年6月20日、CTD 2.7.3.3.2)
44
「IDEC-C2B8視神経脊髄炎スペクトラム障害の再発予防国内臨床試験の概要」(社内資料)
45
「IDEC-C2B8 ABO血液型不適合腎移植国内臨床試験の概要」(承認年月日:2016年2月29日、CTD 2.7.3.2.1)
46
「IDEC-C2B8 ABO血液型不適合腎移植国内臨床試験の概要」(承認年月日:2016年2月29日、CTD 2.7.4.2.1)
47
「IDEC-C2B8 ABO血液型不適合腎移植国内臨床試験の概要」(承認年月日:2016年2月29日、CTD 2.7.4.3.1)
48
「抗ドナー抗体陽性、抗HLA抗体陽性の生体腎移植の抗体関連型拒絶反応抑制国内臨床試験の概要」(CTD 2.7.3.2)
49
Lefaucheur C, et al.:Am J Transplant. 2008;8:324-331
50
Haas M, et al.:Am J Transplant. 2018;18:293-307
51
「抗ドナー抗体陽性、抗HLA抗体陽性の生体腎移植の抗体関連型拒絶反応抑制国内臨床試験の概要」(CTD 2.7.4.2)
52
江川ら.:移植. 2023;58:43-57
53
江川ら.:移植. 2015;50:62-77
54
Akamatsu N, et al.:Transplant Direct. 2021;7:e729
55
「腎移植の抗体関連型拒絶反応治療国内臨床試験の概要」(CTD 2.7.3.2)
56
「腎移植の抗体関連型拒絶反応治療国内臨床試験の概要」(CTD 2.7.4.2)
57
Sakamoto S, et al .:Hepatol Res. 2021;51:990-999
58
縄田ら.:移植. 2021;56:43-52
59
芳川ら.:移植. 2021;56:53-68
60
伊藤ら.:移植. 2021;56:35-42
61
Reff ME, et al.:Blood. 1994;83:435-445
62
「マウス抗体IDEC-2B8のヒトCD20に対する結合特異性」(社内資料)
63
「マウス-ヒトキメラ抗体IDEC-C2B8のヒト末梢血中Bリンパ球及びヒト原発性Bリンパ腫細胞に対する結合特異性」(社内資料)
64
「ヒト正常組織との交叉反応性試験」(承認年月日:2001年6月20日、申請資料概要 ニ. 毒性)

