







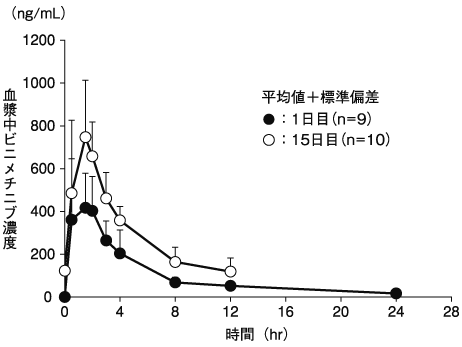


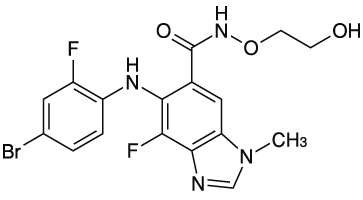
1
小野薬品工業:国内第Ⅰ相(CMEK162X1101)試験成績(社内資料;2019年1月8日承認、CTD2.7.6.8、CTD2.7.2.2.2.2)
2
小野薬品工業:海外第Ⅰ相(CMEK162A2103)試験成績(社内資料;2019年1月8日承認、CTD2.7.6.16)
3
小野薬品工業:In vitro血漿蛋白結合及び血球移行性(社内資料;2019年1月8日承認、CTD2.6.4.4)
4
小野薬品工業:In vitro代謝(社内資料;2019年1月8日承認、CTD2.6.4.5)
5
小野薬品工業:代謝酵素の推定(社内資料;2019年1月8日承認、CTD2.6.4.5)
6
小野薬品工業:海外マスバランス(CMEK162A2102)試験成績(社内資料;2019年1月8日承認、CTD2.7.6.5)
7
小野薬品工業:肝機能障害患者における海外第Ⅰ相(CMEK162A2104)試験成績(社内資料;2019年1月8日承認、CTD2.7.6.12、CTD2.7.2.2.2.2)
8
小野薬品工業:腎機能障害患者における海外第Ⅰ相(ARRAY-162-106)試験成績(社内資料;2019年1月8日承認、CTD2.7.6.11)
9
小野薬品工業:海外第Ⅰ相(ARRAY-162-105)試験成績(社内資料;2019年1月8日承認、CTD2.7.6.15)
10
小野薬品工業:海外第Ⅰ相(CMEK162A2105)試験成績(社内資料;2019年1月8日承認、CTD2.7.6.17、CTD2.7.2.2.2.2)
11
小野薬品工業:CYP阻害作用(社内資料;2019年1月8日承認、CTD2.6.4.5)
12
小野薬品工業:P-gp基質検討(社内資料;2019年1月8日承認、CTD2.6.4.7)
13
小野薬品工業:BCRP基質検討(社内資料;2019年1月8日承認、CTD2.6.4.7)
14
小野薬品工業:国際共同第Ⅲ相(CMEK162B2301)試験成績(社内資料;2019年1月8日承認、CTD2.7.6.19、CTD2.7.6.20)
15
小野薬品工業:国際共同第Ⅲ相(ARRAY-818-302)試験成績(社内資料;2020年11月27日承認、CTD2.7.6.6)
16
小野薬品工業:MEKに対する阻害活性(社内資料;2019年1月8日承認、CTD2.6.2.2)
17
小野薬品工業:ERKのリン酸化に対する阻害作用(社内資料;2019年1月8日承認、CTD2.6.2.2)
18
小野薬品工業:BRAF変異ヒト悪性黒色腫細胞の増殖に対する抑制作用(社内資料;2019年1月8日承認、CTD2.6.2.2)
19
小野薬品工業:BRAF変異ヒト結腸・直腸癌細胞の増殖に対する抑制作用(社内資料;2020年11月27日承認、CTD2.6.2.2)
20
小野薬品工業:BRAF変異ヒト悪性黒色腫細胞の増殖に対するエンコラフェニブとの併用効果(社内資料;2019年1月8日承認、CTD2.6.2.2)
21
小野薬品工業:BRAF変異ヒト結腸・直腸癌細胞の増殖に対するエンコラフェニブとの併用効果(社内資料;2020年11月27日承認、CTD2.6.2.2)
22
小野薬品工業:BRAF変異ヒト悪性黒色腫細胞移植マウスにおける抗腫瘍効果(社内資料;2019年1月8日承認、CTD2.6.2.2)
23
小野薬品工業:BRAF変異ヒト結腸・直腸癌細胞移植マウスにおける抗腫瘍効果(社内資料;2020年11月27日承認、CTD2.6.2.2)
24
小野薬品工業:BRAF変異ヒト悪性黒色腫細胞移植マウスにおけるエンコラフェニブとの併用効果(社内資料;2019年1月8日承認、CTD2.6.2.2)
25
小野薬品工業:BRAF変異ヒト結腸・直腸癌細胞移植マウスにおけるエンコラフェニブ及びセツキシマブとの併用効果(社内資料;2020年11月27日承認、CTD2.6.2.2)

