





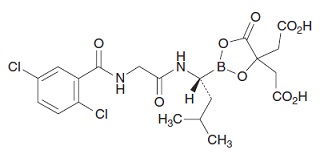
1
イキサゾミブクエン酸エステルの薬物動態試験成績①(2017年3月30日承認, CTD 2.7.6.1)(社内資料)
2
イキサゾミブクエン酸エステルの薬物動態試験成績②(2017年3月30日承認, CTD 2.7.6.7)(社内資料)
3
イキサゾミブクエン酸エステルの薬物動態試験成績③(2017年3月30日承認, CTD 2.7.2)(社内資料)
4
イキサゾミブクエン酸エステルの薬物動態試験成績④(2017年3月30日承認, CTD 2.7.6.9)(社内資料)
5
イキサゾミブクエン酸エステルの非臨床薬物動態試験成績(2017年3月30日承認, CTD 2.6.4.5)(社内資料)
6
イキサゾミブクエン酸エステルの薬物動態試験成績⑤(2017年3月30日承認, CTD 2.7.6.10)(社内資料)
7
イキサゾミブクエン酸エステルの薬物動態試験成績⑥(2017年3月30日承認, CTD 2.7.6.8)(社内資料)
8
イキサゾミブクエン酸エステルの第Ⅲ相試験成績①(2017年3月30日承認, CTD 2.7.6.5)(社内資料)
9
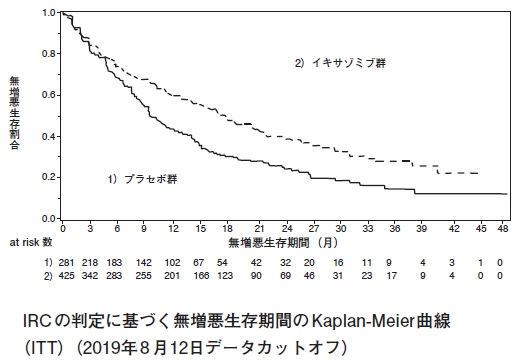
イキサゾミブクエン酸エステルの第Ⅲ相試験成績②(社内資料)
10
イキサゾミブクエン酸エステルの第Ⅲ相試験成績③(社内資料)
11
イキサゾミブクエン酸エステルの非臨床薬理試験成績(2017年3月30日承認, CTD 2.6.2)(社内資料)
12
Chauhan D.et al.: Clin.Cancer Res.2011;17(16): 5311-5321.

