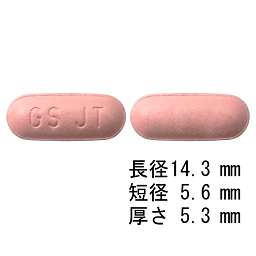
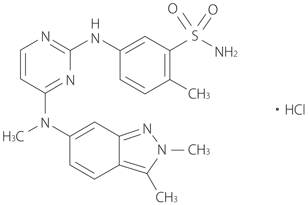
In vitro試験で、本剤の代謝には主にチトクロームP450(CYP)3A4が、一部CYP1A2及び2C8が関与することから、CYP3A4阻害剤及び誘導剤は本剤の薬物動態に影響を及ぼす可能性がある。また、本剤はCYP(2B6、2C8、2E1及び3A4)、UGT1A1及びOATP1B1を阻害し、P-糖蛋白質(Pgp)及びBCRPの基質であった。
本剤(10~100μg/mL)のヒト血漿蛋白結合率はin vitroで99%超であった。また、本剤は主に血清アルブミン(99%超)及びα1-酸性糖蛋白質(96%超)に結合すると考えられた。 外国人固形癌患者3例に14C-標識体400mgを単回経口投与したときの放射能の血液/血漿比は0.59~0.93と血球移行性は低かった。 In vitro試験で、本剤はPgp及びBCRPの基質であった,。
代謝
In vitro試験で、本剤の一部は酸化的に代謝され、この代謝には主にCYP3A4が、一部CYP1A2及び2C8が関与する。 固形癌患者13例に本剤800mg並びに、承認用法・用量(1日1回800mg)とは異なる400及び1000mgを経口投与後の血漿中には、おもに未変化体が検出された。代謝物である脱メチル体及び一酸化体なども検出されたが、個体間のばらつきが大きく、未変化体に対する割合はいずれも5%未満であった。
In vitro試験で、本剤はCYP(1A2、2B6、2C8、2C9、2C19、2D6、2E1及び3A4)及びUGT1A1を阻害し、IC50はそれぞれ7.9~18μM(3.5~7.9μg/mL)及び1.2μM(0.5μg/mL)であった。 外国人固形癌患者24例を対象に本剤がCYP代謝に及ぼす影響について、各種CYPプローブを用いて検討した結果、本剤800mgの1日1回投与ではカフェイン(CYP1A2の基質)、ワルファリン(CYP2C9の基質)及びオメプラゾール(CYP2C19の基質)の薬物動態に意義のある影響を及ぼさなかった。一方、本剤はミダゾラム(CYP3A4の基質)のAUC及びCmaxを約30%増加させ、デキストロメトルファン(CYP2D6の基質)を経口投与後の尿中のデキストロメトルファン/デキストルファン比を33~64%増加させた。 また、in vitro試験で、本剤はOATP1B1を阻害し、IC50は0.79μM(0.3μg/mL)であった。