


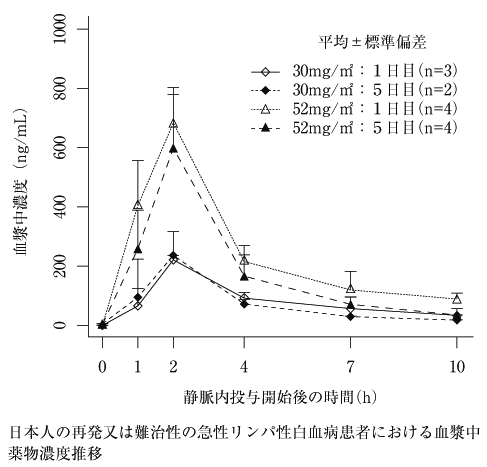
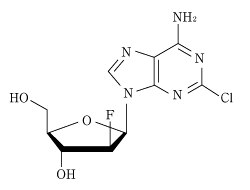
1
社内資料:ラット及びウサギを用いた生殖発生毒性試験(2013年3月25日承認、CTD2.6.6.6)
2
社内資料:細菌を用いたin vitro復帰突然変異試験(2013年3月25日承認、CTD2.6.6.4)
3
社内資料:哺乳動物細胞を用いたin vitro染色体異常試験(2013年3月25日承認、CTD2.6.6.4)
4
社内資料:ラットを用いたin vivo小核試験(2013年3月25日承認、CTD2.6.6.4)
5
社内資料:ラット及びイヌを用いた反復投与毒性試験(2013年3月25日承認、CTD2.6.6.3)
6
社内資料:国内試験:第I相臨床試験(2013年3月25日承認、CTD2.7.6.1)
7
社内資料:in vitro蛋白結合試験(2013年3月25日承認、CTD2.6.4.4)
8
社内資料:in vitro血球移行性試験(2013年3月25日承認、CTD2.6.4.4)
9
社内資料:ヒト単離肝細胞を用いたin vitro CYP450誘導試験(2013年3月25日承認、CTD2.6.4.7)
10
社内資料:ヒト肝ミクロソームを用いたin vitro CYP450阻害試験(2013年3月25日承認、CTD2.6.4.7)
11
社内資料:母集団薬物動態解析(2013年3月25日承認、CTD2.7.2.3)
12
Jeha S, et al.:J Clin Oncol. 2006;24(12):1917-23
13
社内資料:海外試験:第II相臨床試験(2013年3月25日承認、CTD2.7.6.3、2.7.6.5)
14
Xie K C, et al.:Cancer Res. 1996;56(13):3030-7
15
Genini D, et al.:Blood. 2000;96(10):3537-43
16
社内資料:マウスを用いた抗腫瘍効果(2013年3月25日承認、CTD2.6.2.2)

