


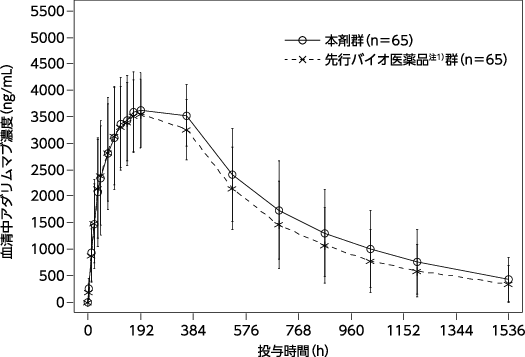
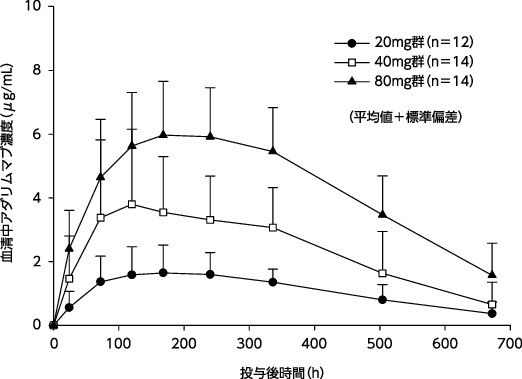
1
Breedveld FC, et al.:Arthritis Rheum. 2006;54:26-37
2
Keystone EC, et al.:Arthritis Rheum. 2004;50:1400-1411
3
van der Heijde D, et al.:Arthritis Rheum. 2006;54:2136-2146
4
Colombel JF, et al.:Gastroenterology 2007;132:52-65
5
Weinblatt ME, et al.:Arthritis Rheum. 2003;48:35-45
6
Furst DE, et al.:J. Rheumatol. 2003;30:2563-2571
7
Gladman DD, et al.:Ann. Rheum. Dis. 2007;66:163-168
8
Gladman DD, et al.:Arthritis Rheum. 2007;56:476-488
9
Hanauer SB, et al.:Gastroenterology 2006;130:323-333
10
Sandborn WJ, et al.:Gut 2007;56:1232-1239
11
社内資料:健康成人を対象とした第Ⅰ相/臨床薬理試験(FKB327-006試験)
12
関節リウマチ患者を対象としたアダリムマブ単回皮下投与時の臨床薬理試験(2008年4月16日承認、ヒュミラ®皮下注 申請資料概要2.7.6.15)
13
健康成人被験者を対象としたアダリムマブの臨床試験用製剤と市販用製剤の非盲検無作為化並行群間生物学的同等性試験(2008年4月16日承認、ヒュミラ®皮下注 申請資料概要2.7.6.3)
14
生物学的利用率・生物学的同等性(2008年4月16日承認、ヒュミラ®皮下注 申請資料概要2.5.1.3及び2.5.2.3)
15
Miyasaka N. The CHANGE Study Investigators:Mod Rheumatol. 2008;18:252-262
16
日本人乾癬患者における薬物動態及び日本人RA患者との比較(2010年1月20日承認、ヒュミラ®皮下注 申請資料概要2.5.3.4)
17
日本人活動性強直性脊椎炎患者(M10-239試験)(2010年10月27日承認、ヒュミラ®皮下注 申請資料概要2.7.2.2)
18
薬物動態(日本人若年性特発性関節炎患者)(2011年7月1日承認、ヒュミラ®皮下注 申請資料概要2.5.3.1)
19
血清中アダリムマブ濃度(日本人腸管型ベーチェット病患者)(2013年5月16日承認、ヒュミラ®皮下注 申請資料概要2.5.3.1)
20
日本人クローン病患者(2010年10月27日承認、ヒュミラ®皮下注 申請資料概要2.7.2.2)
21
国内第Ⅲ相試験(M13-687試験)(2016年6月20日承認、ヒュミラ®皮下注 審査報告書)
22
Suzuki Y, et al.:J Gastroenterol. 2014;49:283-294
23
M14-033試験(2021年9月27日承認、ヒュミラ®皮下注 申請資料概要2.7.2.2)
24
第Ⅲ相試験(M10-877試験、M10-880試験)(2016年9月28日承認、ヒュミラ®皮下注 審査報告書)
25
アダリムマブの薬物動態特性(2008年4月16日承認、ヒュミラ®皮下注 申請資料概要2.5.3.1)
26
薬物動態試験の概要:排泄(2008年4月16日承認、ヒュミラ®皮下注 申請資料概要2.6.4.6)
27
Ben-Horin S, et al.:Clin. Gastroenterol. Hepatol. 2010;8:475-476
28
社内資料:リウマチ患者を対象とした第Ⅲ相臨床試験(FKB327-002試験)
29
国内二重盲検プラセボ対照試験(M02-575)(2008年4月16日承認、ヒュミラ®皮下注 申請資料概要2.7.4.2)
30
Takeuchi T, et al.:Ann. Rheum. Dis. 2014;73:536-543
31
国内第Ⅲ相試験(M06-859試験)(2012年8月10日承認、ヒュミラ®皮下注 審査報告書)
32
van de Putte LBA, et al.:Ann. Rheum. Dis. 2004;63:508-516
33
海外参考DE013試験:実薬(MTX対照)(2008年4月16日承認、ヒュミラ®皮下注 申請資料概要2.7.4.2)
34
海外第Ⅲ相試験(関節破壊の進展防止)(2008年4月6日承認、ヒュミラ®皮下注 申請資料概要2.7.6.27)
35
Asahina A, et al.:J. Dermatol. 2010;37:299-310
36
M04-688試験(2010年1月20日承認、ヒュミラ®皮下注 申請資料概要2.7.4.2)
37
Morita A, et al.:J. Dermatol. 2018;45:1371-1380
38
Mease PJ, et al.:Arthritis Rheum. 2005;52:3279-3289
39
Kobayashi S, et al.:Mod Rheumatol. 2012;22:589-597
40
M10-239試験(2010年10月27日承認、ヒュミラ®皮下注 申請資料概要2.7.4.2)
41
Imagawa T, et al.:Clin Rheumatol. 2012;31:1713-1721
42
国内で実施したJIAを対象とした臨床試験(M10-240)(2011年7月1日承認、ヒュミラ®皮下注 申請資料概要2.7.4.2)
43
Lovell DJ, et al.:N. Engl. J. Med. 2008;359:810-820
44
海外で実施したJIAを対象とした臨床試験(DE038)(2011年7月1日承認、ヒュミラ®皮下注 申請資料概要2.7.4.1及び2.7.4.2)
45
Tanida S, et al.:Clinical Gastroenterol Hepatol. 2015;13:940-948
46
M11-509試験(2013年5月16日承認、ヒュミラ®皮下注 申請資料概要2.7.4.2)
47
Watanabe M, et al.:J. Crohns Colitis 2012;6:160-173
48
国内で実施したクローン病患者を対象とした臨床試験(M04-729試験)(2010年10月27日承認、ヒュミラ®皮下注 申請資料概要2.7.4.2)
49
国内で実施したクローン病患者を対象とした臨床試験(M06-837試験)(2010年10月27日承認、ヒュミラ®皮下注 申請資料概要2.7.4.2)
50
海外第Ⅱ/Ⅲ相試験(M02-403試験)(2010年10月27日承認、ヒュミラ®皮下注 申請資料概要2.7.6.5)
51
Sandborn WJ, et al.:Ann. Intern. Med. 2007;146:829-838
52
海外第Ⅲ相試験(M04-691試験)(2010年10月27日承認、ヒュミラ®皮下注 申請資料概要2.7.6.7)
53
海外第Ⅲ相試験(M02-404試験)(2010年10月27日承認、ヒュミラ®皮下注 申請資料概要2.7.6.6)
54
国内第Ⅱ/Ⅲ相試験(M10-447試験)、海外第Ⅲ相試験(M06-826試験、M06-827試験)(2013年6月14日承認、ヒュミラ®皮下注 審査報告書)
55
Reinisch W, et al.:Gut 2011;60:780-787
56
Sandborn WJ, et al.:Gastroenterology 2012;142:257-265
57
成人UC患者を対象とした第Ⅲ相試験(M14-033試験)(2021年9月27日承認、ヒュミラ®皮下注 審査報告書)
58
M14-033試験(2021年9月27日承認、ヒュミラ®皮下注 申請資料概要2.5.5.5)
59
社内資料:in vitro薬効薬理試験
60
社内資料:in vivo薬効薬理試験
61
効力を裏付ける試験(in vitro試験)(2008年4月16日承認、ヒュミラ®皮下注 審査報告書)
62
Salfeld J, et al.:Arthritis Rheum. 1998;41:S57

