1
Besarab A, et al.: N Engl J Med. 1998; 339: 584-590
2
Singh AK, et al.: N Engl J Med. 2006; 355: 2085-2098
3
Pfeffer MA, et al.: N Engl J Med. 2009; 361: 2019-2032


HIF-PH阻害薬腎性貧血治療薬
1錠 92.9円
有効成分 | 1錠中モリデュスタット 5mg含有 モリデュスタットナトリウムとして 5.35mg |
|---|---|
添加剤 | 結晶セルロース、クロスカルメロースナトリウム、ヒプロメロース、酸化チタン、タルク、乳糖水和物、ステアリン酸マグネシウム、マクロゴール6000 EP/NF、黄色三二酸化鉄 |
有効成分 | 1錠中モリデュスタット 12.5mg含有 モリデュスタットナトリウムとして 13.375mg |
|---|---|
添加剤 | 結晶セルロース、クロスカルメロースナトリウム、ヒプロメロース、酸化チタン、タルク、D-マンニトール、軽質無水ケイ酸、フマル酸ステアリルナトリウム |
有効成分 | 1錠中モリデュスタット 25mg含有 モリデュスタットナトリウムとして 26.75mg |
|---|---|
添加剤 | 結晶セルロース、クロスカルメロースナトリウム、ヒプロメロース、酸化チタン、タルク、D-マンニトール、軽質無水ケイ酸、フマル酸ステアリルナトリウム、三二酸化鉄 |
有効成分 | 1錠中モリデュスタット 75mg含有 モリデュスタットナトリウムとして 80.25mg |
|---|---|
添加剤 | 結晶セルロース、クロスカルメロースナトリウム、ヒプロメロース、酸化チタン、タルク、D-マンニトール、軽質無水ケイ酸、フマル酸ステアリルナトリウム |
| 剤形 | フィルムコーティング錠 |
|---|---|
| 色調 | 淡赤黄色 |
| 外形 | 表面  裏面 側面  |
| 大きさ | 大きさ(直径) 5mm 大きさ(厚さ) 2.9mm |
| 質量 | 61.75mg |
| 剤形 | フィルムコーティング錠 |
|---|---|
| 色調 | 白色 |
| 外形 | 表面  裏面 側面  |
| 大きさ | 大きさ(直径) 5.5mm 大きさ(厚さ) 2.4mm |
| 質量 | 64.70mg |
| 剤形 | フィルムコーティング錠 |
|---|---|
| 色調 | 灰黄赤色 |
| 外形 | 表面  裏面 側面  |
| 大きさ | 大きさ(直径) 7mm 大きさ(厚さ) 3.1mm |
| 質量 | 128.6mg |
| 剤形 | フィルムコーティング錠 |
|---|---|
| 色調 | 白色 |
| 外形 | 表面  裏面 側面  |
| 大きさ | 大きさ(長径) 11mm 大きさ(短径) 5mm 大きさ(厚さ) 4.5mm |
| 質量 | 206.0mg |
本剤投与量(mg) | ダルベポエチン アルファ(μg) | エポエチン ベータ ペゴル(μg) | エポエチン アルファ又はベータ(IU) | ||
2週に1回 | 4週に1回 | 4週に1回 | 週に1回 | 2週に1回 | |
25 | 15以下 | 30以下 | 25以下 | 1500以下 | 3000以下 |
50 | 15超 | 30超 | 25超 | 1500超 | 3000超 |
段階 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
本剤投与量 | 5mg | 12.5mg | 25mg | 50mg | 75mg | 100mg | 150mg | 200mg |
4週間のHb値上昇 | Hb値 | 用量調節 | |
保存期慢性腎臓病患者及び腹膜透析患者 | 血液透析患者 | ||
0.5g/dL未満 | 10.5g/dL未満 | 9.5g/dL未満 | 1段階増量 |
10.5g/dL以上 | 9.5g/dL以上 | 同じ用量を維持 | |
0.5g/dL以上1.0g/dL未満 | すべての値 | ||
1.0g/dL以上2.0g/dL以下 | 11.0g/dL以下 | 10.0g/dL以下 | |
11.0g/dL超 | 10.0g/dL超 | 1段階減量 | |
2.0g/dL超 | すべての値 | ||
用量調節 | 1段階増量 | 同じ用量を維持注1) | 1段階減量 | 休薬注2) | |
Hb値 | 保存期慢性腎臓病患者 及び 腹膜透析患者 | 11.0g/dL未満 | 11.0g/dL以上 12.5g/dL未満 | 12.5g/dL以上 13.0g/dL未満 | 13.0g/dL以上 |
血液透析患者 | 10.0g/dL未満 | 10.0g/dL以上 12.0g/dL未満 | 12.0g/dL以上 13.0g/dL未満 | ||
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
HIVプロテアーゼ阻害剤 アタザナビル、リトナビル、ロピナビル・リトナビル等 チロシンキナーゼ阻害剤 ソラフェニブ、エルロチニブ、ニロチニブ等 トラニラスト | 本剤の作用が増強するおそれがあるため、併用する場合は、本剤の減量を考慮するとともに、患者の状態を慎重に観察すること。 | 本剤をアタザナビルと同時投与したところ、本剤のAUC(0-∞)及びCmaxは上昇した。 UGT1A1阻害により本剤のクリアランスが低下する。 |
多価陽イオン(カルシウム、鉄、マグネシウム、アルミニウム等)を含有する経口製剤 | 本剤の吸収が低下し、効果が減弱するおそれがあるため、併用する場合は、前後1時間以上間隔をあけて本剤を投与すること。 | 本剤を硫酸鉄と同時投与したところ、本剤のAUC(0-∞)及びCmaxは低下した。 本剤の消化管からの吸収が減少し、血中濃度が低下する。 |
1%以上 | 1%未満 | 頻度不明 | |
|---|---|---|---|
代謝および栄養障害 | 鉄欠乏 | ||
精神障害 | 不眠症 | ||
神経系障害 | めまい(浮動性、回転性) | ||
眼障害 | 眼出血、糖尿病網膜症 | 結膜炎、眼瞼炎 | |
心臓障害 | 心のう液貯留 | ||
血管障害 | 高血圧 | 血圧低下 | |
胃腸障害 | 便秘、下痢、悪心、嘔吐、腹痛 | ||
皮膚および皮下組織障害 | 発疹、そう痒症 | ||
一般・全身障害および投与部位の状態 | 浮腫 |
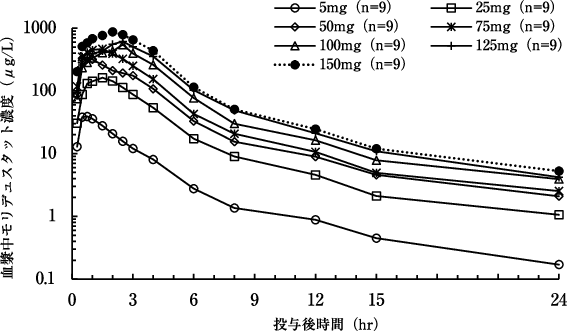
投与量 | n | AUC(0-∞) (μg・h/L) | Cmax (μg/L) | tmax※ (h) | t1/2 (h) |
5mg | 9 | 112 (11.6) | 57.7 (28.5) | 0.50 (0.50-1.50) | 5.57 (24.0) |
25mg | 9 | 614 (19.4) | 226 (28.0) | 1.00 (0.50-2.50) | 8.80 (38.5) |
50mg | 9 | 1230 (11.2) | 511 (45.9) | 0.50 (0.50-3.00) | 9.46 (24.2) |
75mg | 9 | 1710 (23.0) | 640 (32.5) | 0.75 (0.50-1.50) | 9.69 (35.8) |
100mg | 9 | 2510 (20.8) | 988 (49.2) | 2.00 (0.50-4.00) | 7.05 (41.8) |
125mg | 9 | 3020 (21.7) | 1100 (27.9) | 1.50 (0.50-4.00) | 9.41 (39.3) |
150mg | 9 | 3780 (17.6) | 1220 (21.4) | 2.00 (0.25-4.00) | 8.75 (25.0) |
投与量 | n | AUC(0-24)ss (μg・h/L) | Cmax,ss (μg/L) | tmax,ss※ (h) | t1/2,ss (h) |
5mg | 9 | 120 (11.9) | 55.6 (50.6) | 0.50 (0.25-2.00) | 6.25 (37.4) |
25mg | 9 | 657 (19.6) | 319 (40.0) | 1.00 (0.50-2.50) | 6.20 (54.1) |
50mg | 9 | 1260 (16.0) | 606 (31.4) | 0.75 (0.50-2.50) | 7.87 (66.0) |
75mg | 9 | 2030 (28.3) | 698 (41.2) | 2.00 (0.50-2.00) | 6.52 (32.1) |
100mg | 9 | 2530 (21.5) | 1008 (55.5) | 1.50 (0.50-3.00) | 7.45 (30.0) |
125mg | 9 | 3140 (22.1) | 953 (29.5) | 2.00 (0.50-2.50) | 8.26 (42.4) |
150mg | 9 | 4030 (13.3) | 1150 (14.0) | 2.50 (2.00-3.00) | 6.90 (25.8) |
併用薬 | 併用薬 投与量 | 本剤 投与量 | 例数 | 幾何平均値の比[90%信頼区間] (アタザナビル併用投与時/本剤単独投与時) | |
Cmax | AUC(0-∞) | ||||
アタザナビル | 400mg | 25mg | 13 | 2.07 [1.53、2.79] | 2.07 [1.87、2.29] |
併用薬 | 併用薬 投与量 | 本剤 投与量 | 投与条件 | 例数 | 幾何平均値の比[90%信頼区間] (硫酸鉄又は硫酸グリシン併用投与時/本剤単独投与時) | |
Cmax | AUC(0-∞) | |||||
硫酸鉄 | 304mg | 150mg | 同時投与 | 14注1) | 0.16 [0.12、0.22] | 0.25 [0.19、0.33] |
硫酸鉄投与4時間後に本剤投与 | 14 | 0.90 [0.64、1.26] | 0.91 [0.77、1.07] | |||
硫酸鉄投与2時間後に本剤投与 | 14 | 1.00 [0.71、1.40] | 0.84 [0.71、0.99] | |||
硫酸鉄投与1時間前に本剤投与 | 14 | 0.88 [0.63、1.23] | 0.74 [0.63、0.88] | |||
硫酸鉄グリシン | 567.7mg | 同時投与 | 14 | 0.54 [0.39、0.76] | 0.50 [0.42、0.60] | |
併用薬 | 併用薬 投与量 | 本剤 投与量 | 投与条件 | 例数 | 幾何平均値の比[90%信頼区間] (硫酸鉄併用投与時/本剤単独投与時) | |
Cmax | AUC(0-∞) | |||||
硫酸鉄 | 304mg | 150mg | 同時投与 | 11注2) | 0.39 [0.29、0.51] | 0.49 [0.42、0.58] |
硫酸鉄投与1時間後に本剤投与 | 11 | 0.80 [0.60、1.06] | 0.80 [0.70、0.93] | |||
硫酸鉄投与1時間前に本剤投与 | 11注3) | 0.60 [0.45、0.80] | 0.66 [0.57、0.77] | |||
併用薬 | 併用薬 投与量 | 本剤 投与量 | 例数 | 幾何平均値の比[90%信頼区間] (酢酸カルシウム併用投与時/本剤単独投与時) | |
Cmax | AUC(0-∞) | ||||
酢酸カルシウム | 1900mg | 150mg | 15 | 0.53 [0.39、0.72] | 0.85 [0.67、1.09] |
併用薬 | 併用薬 投与量 | 本剤 投与量 | 例数 | 幾何平均値の比[90%信頼区間] (酸化アルミニウム・水酸化マグネシウム配合剤併用投与時/本剤単独投与時) | |
Cmax | AUC(0-∞) | ||||
酸化アルミニウム・水酸化マグネシウム配合剤 | 900mg・600mg | 50mg | 12 | 0.36 [0.26、0.51] | 0.67 [0.60、0.75] |
併用薬 | 併用薬 投与量 | 本剤 投与量 | 例数 | 幾何平均値の比[90%信頼区間] (オメプラゾール併用投与時/本剤単独投与時) | |
Cmax | AUC(0-∞) | ||||
オメプラゾール | 40mg | 50mg | 12 | 0.96 [0.68、1.35] | 0.95 [0.85、1.07] |
併用薬 | 併用薬 投与量 | 本剤 投与量 | 投与条件 | 例数 | 幾何平均値の比[90%信頼区間] (ロスバスタチン併用投与時/本剤単独投与時) | |
Cmax | AUC(0-∞) | |||||
ロスバスタチン | 5mg | 150mg | 同時投与 | 15注4) | 1.30 [1.11、1.52] | 1.11 [0.96、1.28] |
本剤投与3時間前にロスバスタチン投与 | 15注4) | 0.98 [0.83、1.14] | 0.95 [0.82、1.09] | |||
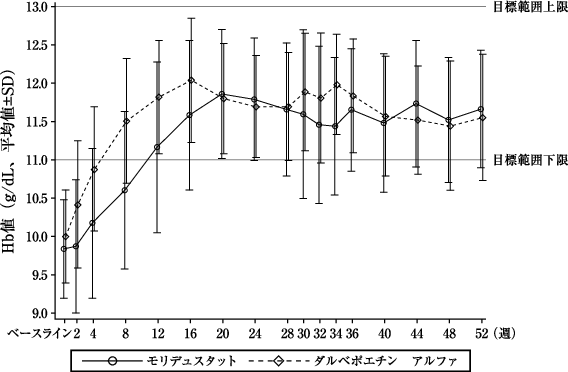
本剤群 (82例) | ダルベポエチン アルファ群(80例) | |
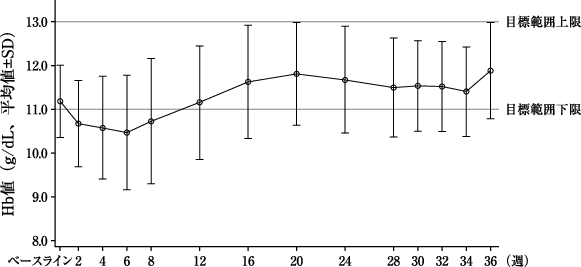
評価期間中(投与30~36週)の平均Hb値[両側95%信頼区間](g/dL)※1 | 11.28±0.98 [11.07、11.50] | 11.70±0.90 [11.50、11.90] |
ベースラインからの変化量(g/dL)※2 | 1.45±1.08 | 1.70±1.03 |
変化量の群間差[両側95%信頼区間](g/dL) | -0.38[-0.67、-0.08] | |
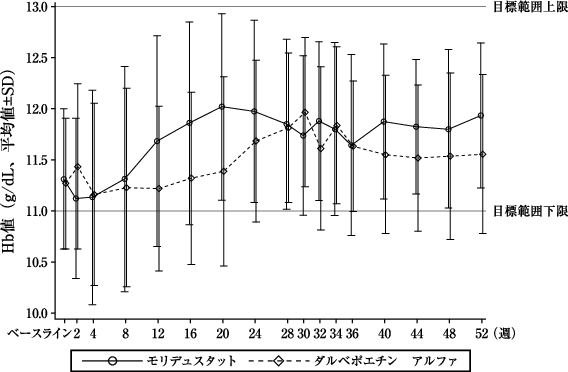
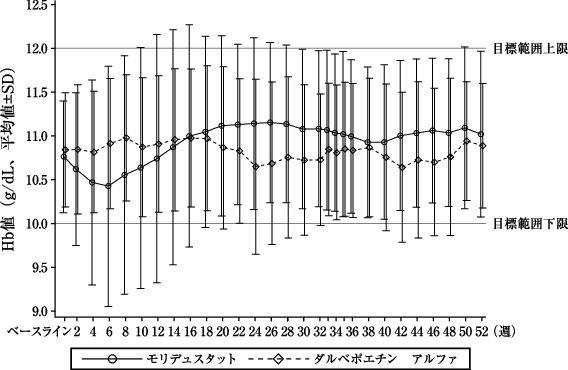
本剤群 (82例) | ダルベポエチン アルファ群(82例) | |
評価期間中(投与30~36週)の平均Hb値[両側95%信頼区間](g/dL)※1 | 11.67±0.83 [11.48、11.85] | 11.53±0.99 [11.31、11.74] |
ベースラインからの変化量(g/dL)※2 | 0.35±1.03 | 0.26±1.00 |
変化量の群間差[両側95%信頼区間](g/dL) | 0.13[-0.15、0.40] | |
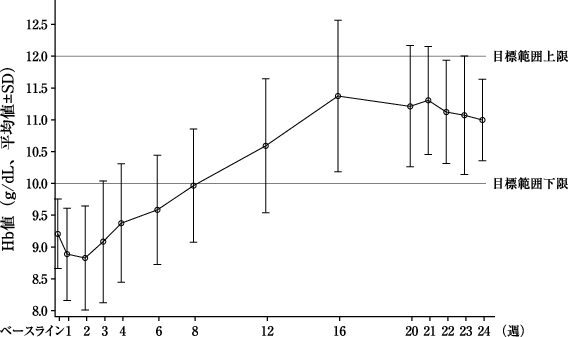
本剤群 (153例) | ダルベポエチン アルファ群(76例) | |
評価期間中(投与33~36週)の平均Hb値[両側95%信頼区間](g/dL)※1 | 10.63±1.34 [10.42、10.84] | 10.77±0.78 [10.59、10.95] |
ベースラインからの変化量(g/dL)※2 | -0.14±1.43 | -0.07±1.00 |
変化量の群間差[両側95%信頼区間](g/dL) | -0.13[-0.46、0.19] | |