


1
社内資料: ミリキズマブの毒性試験(2023年3月27日承認、CTD2.6.6.6.1)
2
Biancone L, et al.: J. Crohns. Colitis. 2016; 10(8): 913-924
3
Taborelli M, et al.: PLoS One. 2020; 15(6): e0235142
4
van den Heuvel TR, et al.: Int. J. Cancer. 2016; 139(6): 1270-1280
5
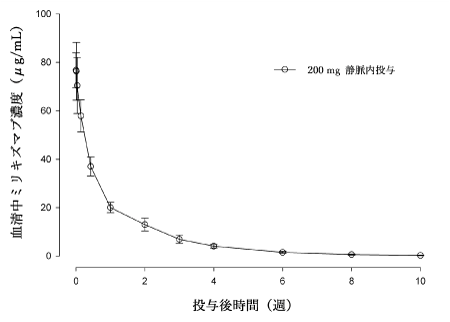
社内資料: 日本人及び外国人健康成人を対象としたミリキズマブの第I相試験(2023年3月27日承認、CTD2.7.2.2.1.1.2)
6
社内資料: ミリキズマブの母集団薬物動態解析(2023年3月27日承認、CTD2.7.2.3)
7
社内資料: 潰瘍性大腸炎患者を対象とした国際共同第III相試験(AMAN試験: 寛解導入療法)(2023年3月27日承認、CTD2.7.6.8)
8
社内資料: 潰瘍性大腸炎患者を対象とした国際共同第III相試験(AMBG試験: 維持療法)(2023年3月27日承認、CTD2.7.6.9)
9
社内資料: ミリキズマブの薬理試験(2023年3月27日承認、CTD2.6.2)

