


1
Lahat A, et al.:J Crohns Colitis.2018 ; 12(1) : 120-123.
2
Julsgaard M, et al.:Gastroenterology.2018 ; 154(3) : 752-754.
3
ベドリズマブの薬力学試験成績(2018年7月2日承認、CTD 2.7.6.8)
4
ベドリズマブの薬物動態試験成績①(2018年7月2日承認、CTD 2.7.2.2)
5
ベドリズマブの潰瘍性大腸炎患者を対象とした国内第Ⅲ相臨床試験成績(2018年7月2日承認、CTD 2.7.6.9)
6
ベドリズマブのクローン病患者を対象とした国内第Ⅲ相臨床試験成績(2019年5月22日承認、CTD 2.7.6.2)
7
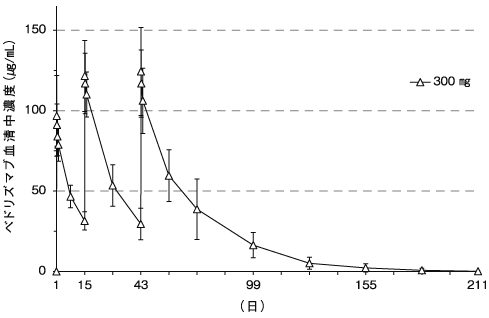
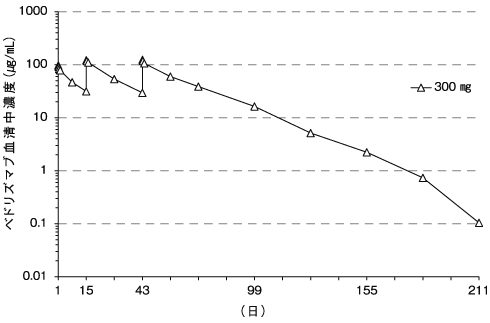
ベドリズマブの薬物動態試験成績②(2018年7月2日承認、CTD 2.7.2.2)
8
ベドリズマブの潰瘍性大腸炎患者を対象とした海外第Ⅲ相臨床試験成績(2018年7月2日承認、CTD 2.7.6.10)
9
ベドリズマブのクローン病患者を対象とした海外第Ⅲ相臨床試験成績(2019年5月22日承認、CTD 2.7.6.3)
10
Soler D, et al.:J Pharmacol Exp Ther.2009 ; 330(3) : 864-875.
11
Hesterberg PE, et al.: Gastroenterology.1996 ; 111(5) : 1373-1380.
12
Fedyk ER, et al.:Inflamm Bowel Dis.2012 ; 18(11) : 2107-2119.

