




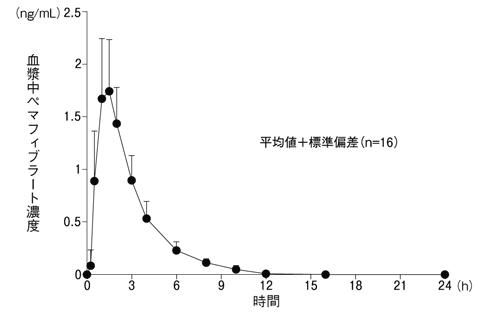
1
興和(株)社内資料: 第Ⅲ相食事の影響試験(2017年7月3日承認、CTD2.7.6.12)
2
興和(株)社内資料: 第Ⅰ相反復投与試験(2017年7月3日承認、CTD2.7.6.3)
3
興和(株)社内資料: 第Ⅰ相マスバランス試験(海外)(2017年7月3日承認、CTD2.7.6.1)
4
興和(株)社内資料: 非臨床試験 薬物動態試験(2017年7月3日承認、CTD2.6.4.1-10)
5
興和(株)社内資料: 第Ⅲ相腎機能障害患者を対象とした薬物動態試験(2017年7月3日承認、CTD2.7.6.6)
6
興和(株)社内資料: 腎機能障害患者を対象とした製造販売後臨床試験
7
興和(株)社内資料: 第Ⅲ相肝機能障害患者を対象とした薬物動態試験(2017年7月3日承認、CTD2.7.6.5)
8
興和(株)社内資料: 薬物相互作用試験①(海外)(2017年7月3日承認、CTD2.7.6.13-17、2.7.6.29-30)
9
興和(株)社内資料: 薬物相互作用試験②(海外を含む)(2017年7月3日承認、CTD2.7.6.8-11)
10
興和(株)社内資料: 第Ⅱ/Ⅲ相フェノフィブラートとの比較検証試験(2017年7月3日承認、CTD2.7.6.22)
11
興和(株)社内資料: 第Ⅲ相フェノフィブラートとの比較検証試験(2017年7月3日承認、CTD2.7.6.24)
12
興和(株)社内資料: 第Ⅲ相TG高値を示す脂質異常症患者を対象とした52週長期投与試験(2017年7月3日承認、CTD2.7.6.28)
13
興和(株)社内資料: 第Ⅲ相2型糖尿病を合併した脂質異常症患者を対象とした長期投与試験(2017年7月3日承認、CTD2.7.6.26)
14
Fruchart JC.: Cardiovasc Diabetol. 2013; 12: 82.
15
Sahebkar A, et al.: Expert Opin Pharmacother. 2014; 15: 493-503.
16
Pawlak M, et al.: J Hepatol. 2015; 62: 720-33.
17
興和(株)社内資料: 非臨床試験 薬理試験(2017年7月3日承認、CTD2.6.2.1-8)

