




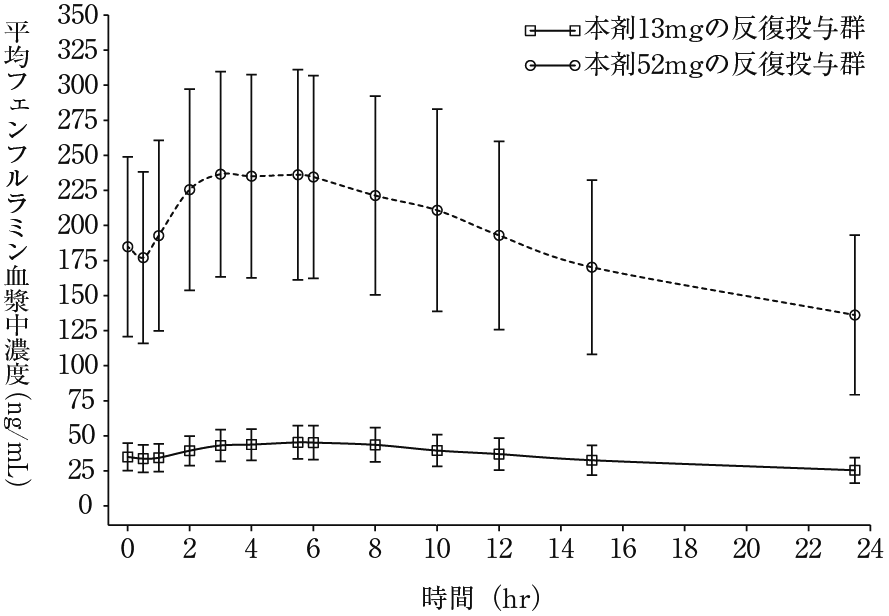
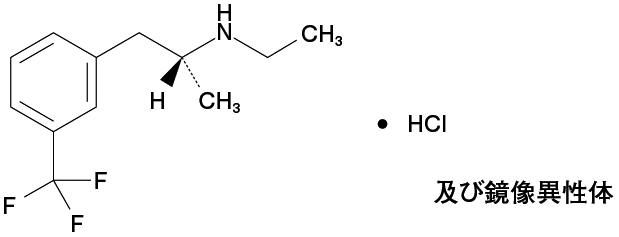
1
社内資料:日本人及び白人健康成人を対象とした薬物動態試験(2022年9月26日承認、CTD 2.7.2.2.1.1)
2
社内資料:健康成人における反復投与薬物動態(2022年9月26日承認、CTD 2.7.2.2.1.5)
3
社内資料:母集団薬物動態モデルの検討(2022年9月26日承認、CTD 2.7.2.2.5.1)
4
社内資料:薬物動態を含む第III相臨床試験(2022年9月26日承認、CTD 2.7.2.2.2.2)
5
社内資料:薬物動態を含む第III相臨床試験(2022年9月26日承認、CTD 2.7.2.2.2.3)
6
社内資料:薬物動態を含む第III相臨床試験
7
社内資料:薬物相互作用及び食事の影響(2022年9月26日承認、CTD 2.7.2.2.1.2)
8
社内資料:In vitroにおける蛋白結合に関する検討(2022年9月26日承認、CTD 2.7.2.2.3.1)
9
社内資料:In vitroにおける遺伝子組み換え酵素に関する検討(2022年9月26日承認、CTD 2.7.2.2.3.6)
10
社内資料:In vitroにおけるCYP表現型別の代謝に関する検討(2022年9月26日承認、CTD 2.7.2.2.3.3)
11
Marchant NC, et al.: Xenobiotica. 1992; 22(11): 1251-1266
12
Bruce RB, et al.: J Pharm Sci. 1968; 57(7): 1173-1176
13
Beckett AH, et al.: J Pharm Pharmacol. 1967; 19(Suppl): 42S-49S
14
社内資料:腎機能障害患者における薬物動態の検討(2022年9月26日承認、CTD 2.7.2.2.1.6)
15
社内資料:肝機能障害患者における薬物動態の検討(2022年9月26日承認、CTD 2.7.2.2.1.7)
16
社内資料:健康被験者を対象とした薬物動態試験(2022年9月26日承認、CTD 2.7.2.2.1.4)
17
社内資料:国際共同第III相臨床試験(2022年9月26日承認、CTD 2.7.6.10)
18
社内資料:国際共同第III相臨床試験(2022年9月26日承認、CTD 2.7.6.8)
19
社内資料:国際共同第III相臨床試験(2022年9月26日承認、CTD 2.7.6.9)
20
社内資料:国際共同第III相臨床試験
21
Hekmatpanah CR, et al.: Eur J Pharmacol. 1990; 177(1-2): 95-98
22
Rothman RB, et al.: Circulation. 2000; 102(23): 2836-2841
23
Sourbron J, et al.: ACS Chem Neurosci. 2016; 7(5): 588-598
24
Wong JC, et al.: FASEB J. 2017; 31(suppl 1): 813.7
25
Rodríguez-Muñoz M, et al.: Oncotarget. 2018; 9(34): 23373-23389
26
Buterbaugh GG: Life Sci. 1978; 23(24): 2393-2404
27
Lazarova M, et al.: Life Sci. 1983; 32(20): 2343-2348

