添付文書番号
6343449D1024_1_04
企業コード
100806
作成又は改訂年月
日本標準商品分類番号
876343
薬効分類名
血漿分画製剤(静注用人プロトロンビン複合体製剤)
承認等
販売名
ケイセントラ静注用500
販売名コード
販売名英字表記
Kcentra for I.V. Injection 500
販売名ひらがな
けいせんとらじょうちゅうよう
承認番号等
販売開始年月
貯法、有効期間
貯法
凍結を避けて25℃以下で保存
有効期間
製造日から36箇月
基準名
規制区分
販売名
ケイセントラ静注用1000
販売名コード
販売名英字表記
Kcentra for I.V. Injection 1000
販売名ひらがな
けいせんとらじょうちゅうよう
承認番号等
販売開始年月
貯法、有効期間
貯法
凍結を避けて25℃以下で保存
有効期間
製造日から36箇月
基準名
規制区分
一般的名称
乾燥濃縮人プロトロンビン複合体
特殊記載項目
本剤は、貴重なヒト血液を原料等として製剤化したものである。原料となった血液を採取する際には、問診、感染症関連の検査を実施するとともに、製造工程における一定の不活化・除去処理などを実施し、感染症に対する安全対策を講じているが、ヒト血液を原料としていることによる感染症伝播のリスクを完全に排除することはできないため、疾病の治療上の必要性を十分に検討の上、必要最小限の使用にとどめること。
禁忌(次の患者には投与しないこと)
播種性血管内凝固(DIC)状態の患者[過凝固状態を誘発又は悪化させる可能性がある。]
組成・性状
組成
ケイセントラ静注用500
有効成分 | 人プロトロンビン複合体※注1),注2) 500IU |
|---|
添加剤 | アンチトロンビンIII※※ 4-30IU 人血清アルブミン※※ 40-80mg ヘパリンナトリウム 8-40IU 塩化ナトリウム 60-120mg クエン酸ナトリウム水和物 45-92mg 塩酸 適量 水酸化ナトリウム 適量 |
|---|
添付溶解液
(日局注射用水) | 20mL |
|---|
| 備考(※) | ヒト血液由来成分
採血国:ドイツ
採血の区分注3) :献血
及び
採血国:米国
採血の区分注3) :非献血 |
|---|
| 備考(※※) | ヒト血液由来成分
採血国:米国
採血の区分注3) :非献血 |
|---|
ケイセントラ静注用1000
有効成分 | 人プロトロンビン複合体※注1),注2) 1,000IU |
|---|
添加剤 | アンチトロンビンIII※※ 8-60IU 人血清アルブミン※※ 80-160mg ヘパリンナトリウム 16-80IU 塩化ナトリウム 120-240mg クエン酸ナトリウム水和物 91-183mg 塩酸 適量 水酸化ナトリウム 適量 |
|---|
添付溶解液
(日局注射用水) | 40mL |
|---|
| 備考(※) | ヒト血液由来成分
採血国:ドイツ
採血の区分注3) :献血
及び
採血国:米国
採血の区分注3) :非献血 |
|---|
| 備考(※※) | ヒト血液由来成分
採血国:米国
採血の区分注3) :非献血 |
|---|
本剤は製造工程でアンチトロンビンIII(採血国:米国、採血の区分注3) :非献血)及びヘパリンナトリウム(ブタの腸粘膜由来成分)を使用している。
IU:国際単位
製剤の性状
ケイセントラ静注用500
| pH | 6.5~7.5 |
|---|
| 浸透圧比 | 約0.6(生理食塩液に対する比) |
|---|
| 性状 | 白色又はわずかに着色した粉末又はもろい塊であり、注射用水で溶解するとき、無色ないし淡黄色のほとんど澄明な液となる。 |
|---|
ケイセントラ静注用1000
| pH | 6.5~7.5 |
|---|
| 浸透圧比 | 約0.6(生理食塩液に対する比) |
|---|
| 性状 | 白色又はわずかに着色した粉末又はもろい塊であり、注射用水で溶解するとき、無色ないし淡黄色のほとんど澄明な液となる。 |
|---|
効能又は効果
ビタミンK拮抗薬投与中の患者における、急性重篤出血時、又は重大な出血が予想される緊急を要する手術・処置の施行時の出血傾向の抑制
用法及び用量
通常、血液凝固第IX因子として、下記の投与量を単回静脈内投与する。
投与前のプロトロンビン時間-国際標準比(PT-INR) | 投与量 |
体重100kg以下の場合 | 体重100kgを超える場合 |
2~<4 | 25IU/kg | 2500IU |
4~6 | 35IU/kg | 3500IU |
>6 | 50IU/kg | 5000IU |
用法及び用量に関連する注意
本剤の投与を受ける患者には、ビタミンK製剤の併用を考慮すること。
重要な基本的注意
本剤の使用にあたっては、疾病の治療における本剤の必要性とともに、本剤の製造に際し感染症の伝播を防止するための安全対策が講じられているが、血液を原料等としていることに由来する感染症伝播のリスクを完全に排除することができないことを患者に対して説明し、理解を得るよう努めること。
本剤の原料等となる血漿については、HBs抗原、抗HCV抗体、抗HIV-1抗体及び抗HIV-2抗体が陰性であることを確認している。さらに、プールした試験血漿については、HIV-1、HBV、HCV及びHAVについて核酸増幅検査(NAT)を実施し、検出限界以下であることを確認している。また、ヒトパルボウイルスB19についてもNATを実施し、一定の基準に適合した血漿を用いている。
その後の製造工程では、ウイルス除去や不活化の工程として60℃10時間の液状加熱処理、硫酸アンモニウム沈殿/リン酸カルシウム吸着及びナノフィルトレーションによる処理を実施しているが、現在の製造工程では、ヒトパルボウイルスB19等のウイルスを完全に不活化・除去することが困難である。そのため、本剤の投与によりその感染の可能性を否定できないので、投与後の経過を十分に観察すること。
現在までに本剤の投与により変異型クロイツフェルト・ヤコブ病(vCJD)等が伝播したとの報告はない。しかしながら、製造工程において異常プリオンを低減し得るとの報告があるものの、理論的なvCJD等の伝播のリスクを完全には排除できないので、投与の際には患者への説明を十分行い、治療上の必要性を十分検討の上投与すること。
本剤の投与は、出血性及び血栓性疾患に関する十分な知識・治療経験を有する医師のもとで行うこと。
本剤の効果を確認するため、必要に応じ血液凝固能のモニタリングを行うこと。十分な効果が得られない場合には、患者の状態に応じ、他の適切な治療を行うこと。本剤の追加投与に対する有効性及び安全性は検討されていない。また、本剤の追加投与後に血栓塞栓性事象を発現し、死亡した症例が報告されている。
止血後は、患者の状態を十分に観察し、血栓塞栓症の発現リスクと出血リスクを考慮した上で、抗凝固剤の再開を検討すること。
本剤には添加剤としてヘパリンが含まれているため、ヘパリン起因性血小板減少症(HIT:heparin-induced thrombocytopenia)があらわれる可能性がある。本剤投与後に血小板数を測定し、血小板の著明な減少がみられた場合には、適切な処置を行うこと。
特定の背景を有する患者に関する注意
合併症・既往歴等のある患者
本剤の成分に対し過敏症の既往歴のある患者
ヘパリン起因性血小板減少症(HIT:heparin-induced thrombocytopenia)の既往歴のある患者
溶血性・失血性貧血の患者
ヒトパルボウイルスB19の感染を起こす可能性を否定できない。感染した場合には、発熱と急激な貧血を伴う重篤な全身症状を起こすことがある。
免疫不全患者・免疫抑制状態の患者
ヒトパルボウイルスB19の感染を起こす可能性を否定できない。感染した場合には、持続性の貧血を起こすことがある。
妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。本剤の投与によりヒトパルボウイルスB19の感染の可能性を否定できない。感染した場合には胎児への障害(流産、胎児水腫、胎児死亡)が起こる可能性がある。
小児等
低出生体重児、新生児、乳児、幼児又は小児には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。小児等を対象とした有効性及び安全性を指標とした臨床試験は実施していない。
高齢者
患者の状態を観察しながら慎重に投与すること。一般に生理機能が低下している。
副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
重大な副作用
血栓塞栓症(3.8%)
血栓塞栓症(致死的な転帰の症例を含む)があらわれることがある。
ショック、アナフィラキシー(頻度不明)
播種性血管内凝固(DIC)(頻度不明)
その他の副作用
| 0.5%以上 | 頻度不明 |
|---|
一般・全身障害及び投与部位の状態 | | 体温上昇 |
|---|
神経系障害 | 頭痛 | |
|---|
免疫系障害 | | 抗体産生、過敏症/アレルギー反応 |
|---|
注)副作用頻度は、海外臨床試験データに基づく。
過量投与
過量投与により、心筋梗塞、DIC、静脈血栓症及び肺塞栓症等を発症する可能性がある。
適用上の注意
薬剤調製時の注意
「ケイセントラ静注用500/1000の使用方法」に従い調製を行うこと。
添付の溶剤以外は使用しないこと。
他の製剤との混注を避けること。
本剤は溶解後ただちに使用すること。
使用後の残液は細菌汚染のおそれがあるので使用しないこと。
薬剤投与時の注意
不溶物又は混濁が認められるものは使用しないこと。
注入速度は3IU/kg/分以下とし、210IU/分を超えないこと。臨床試験において検討されていない。
薬物動態
血中濃度
健康成人(15例)を対象に、本剤50IU/kgを単回静脈内投与した際の薬物動態パラメーターは以下のとおりであった(外国人データ)。
| 血漿中半減期
(時間) | 回収率注1)
(IU/dL)/(IU/kg) |
中央値(範囲) | 幾何平均(90%信頼区間) |
第II因子 | 59.7(25.0-135.3)注2) | 2.2(2.0-2.3) |
第VII因子 | 4.2(2.1-9.2) | 2.4(2.3-2.6) |
第IX因子 | 16.7(9.5-127.1)注2) | 1.6(1.4-1.8) |
第X因子 | 30.7(16.9-43.8) | 2.1(2.0-2.3) |
プロテインC | 47.2(9.3-121.7)注2) | 2.8(2.7-3.0) |
プロテインS | 49.1(33.1-83.3)注2) | 2.0(1.8-2.1) |
臨床成績
有効性及び安全性に関する試験
国内第III相臨床試験(3004試験)
ビタミンK拮抗薬投与中に急性重篤出血を呈した患者(出血群)6例及び緊急の外科手術/侵襲的処置を要する患者(手術群)5例に、投与前のPT-INRに基づき本剤25~50IU/kgを投与した。投与前のPT-INRの中央値(最大値、最小値)は、出血群で4.76(2.26、10.56)、手術群で3.13(2.11、5.82)であった。投与終了後30分時点のPT-INRの中央値(最大値、最小値)は、出血群で1.11(1.01、1.38)、手術群で1.25(1.07、1.92)であった。
副作用は安全性解析対象集団11例中手術群の2例(18.2%)で心房血栓症、脾臓梗塞各1例が認められた。
海外第III相臨床試験(3002試験)
ビタミンK拮抗薬投与中に急性重篤出血を呈した患者に、投与前のPT-INRに基づき本剤25~50IU/kg又は血漿10~15mL/kgを投与した。その結果、主要評価項目において、本剤の血漿に対する非劣性が示された(非劣性限界値:-10%)。
主要評価項目 | 被験者数(%)注1)
[95%信頼区間] | 投与群間差
(本剤-血漿)
%[95%信頼区間] |
本剤
(98例) | 血漿
(104例) |
止血効果が「有効」とされた割合 | 71(72.4%)
[63.6; 81.3] | 68(65.4%)
[56.2; 74.5] | 7.1%
[-5.8; 19.9] |
投与終了後30分の時点でPT-INRが1.3以下に低下した割合 | 61(62.2%)
[52.6; 71.8] | 10(9.6%)
[3.9; 15.3] | 52.6%
[39.4; 65.9] |
副作用は安全性解析対象集団103例中10例(9.7%)に認められ、主な副作用はPT-INR上昇2例(1.9%)であった。また、重篤な副作用は虚血性脳卒中及び深部静脈血栓症各1例(1.0%)であった。
海外第III相臨床試験(3003試験)
ビタミンK拮抗薬投与中に緊急の外科手術/侵襲的処置を要する患者に、投与前のPT-INRに基づき本剤25~50IU/kg又は血漿10~15mL/kgを投与した。その結果、主要評価項目において、本剤の血漿に対する非劣性が示された(非劣性限界値:-10%)。
主要評価項目 | 被験者数(%)注2)
[95%信頼区間] | 投与群間差
(本剤-血漿)
%[95%信頼区間] |
本剤
(87例) | 血漿
(81例) |
止血効果が「有効」とされた割合 | 78(89.7%)
[83.3; 96.1] | 61(75.3%)
[65.9; 84.7] | 14.3%
[2.8; 25.8] |
投与終了後30分の時点でPT-INRが1.3以下に低下した割合 | 48(55.2%)
[44.7; 65.6] | 8(9.9%)
[3.4; 16.4] | 45.3%
[31.9; 56.4] |
副作用は安全性解析対象集団88例中8例(9.1%)に認められ、重篤な副作用は虚血性脳卒中、深部静脈血栓症、血栓症及び末梢静脈疾患各1例(1.1%)であった。
薬効薬理
作用機序
血液凝固第II、第VII、第IX及び第X因子は肝臓でビタミンKの存在下で生合成される血液凝固因子である。本剤は、ビタミンK拮抗薬の投与により減少したこれらの因子を補充することにより出血傾向を抑制する。
出血傾向の抑制作用
クマリン系のビタミンK拮抗薬であるフェンプロクモンを投与されたラットモデル(in vivo)及びワルファリン投与を受けたヒトの血漿において、本剤はプロトロンビン時間(PT)を低下させた。また、トロンビン生成試験において、トロンビン濃度のピーク値を上昇させた 。
ラットのフェンプロクモン誘発性出血モデル(in vivo)において、本剤50IU/kg投与によりPT及び活性化部分トロンボプラスチン時間(aPTT)は正常化され、クマリン過量投与による出血を防いだ 。
取扱い上の注意
外箱開封後は遮光して保存すること。
本剤は特定生物由来製品に該当することから、本剤を投与又は処方した場合は、医薬品名(販売名)、その製造番号(ロット番号)、投与又は処方した日、投与又は処方を受けた患者の氏名、住所等を記録し、使用日から少なくとも20年間保存すること。
承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
包装
ケイセントラ静注用500 1バイアル
(日局注射用水20mL×1バイアル、薬液用両刃針添付)
ケイセントラ静注用1000 1バイアル
(日局注射用水40mL×1バイアル、薬液用両刃針添付)
主要文献
1
Tanaka, K.A., et al.: Thromb Res. 122, 117, 2008
2
Dickneite, G.: Thromb Res. 119, 643, 2007
文献請求先及び問い合わせ先
〒107-0061 東京都港区北青山一丁目2番3号
製造販売業者等
CSLベーリング株式会社
東京都港区北青山一丁目2番3号
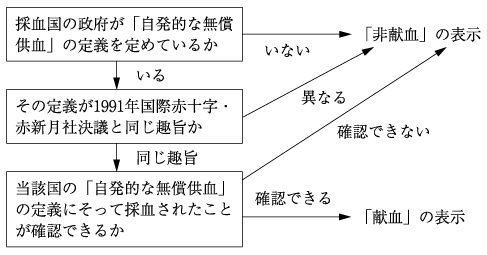
[献血又は非献血の区別の考え方]
献血又は非献血の区別は製剤の安全性の優劣を示すものではありません。この表示区別は、下記の手順に従って決められています。
[ケイセントラ静注用500/1000の使用方法]
1. 薬剤バイアル及び溶解液バイアルを室温に戻す。両バイアルのプラスチックキャップをはずし、ゴム栓をアルコール綿等で消毒する。
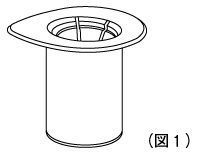
2. 溶解器(薬液用両刃針)のシールを完全にはがして開封する。ブリスター包装から取り出さないこと。(図1)
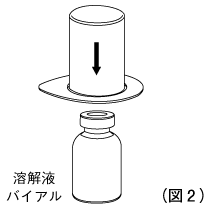
3. 溶解液バイアルを水平の台に置き、しっかりと握る。溶解器をブリスター包装に入れたままの状態で取り、青色側アダプターの穿刺部を、溶解液バイアルのゴム栓にまっすぐ下向きに刺しこむ。(図2)
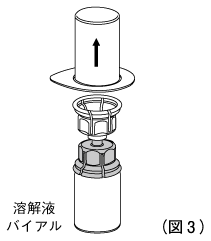
4. ブリスター包装の縁をつかみ、ブリスター包装のみを垂直に引き上げ、溶解器から慎重に取り外す。このとき、溶解器を一緒に引き上げないよう注意する。(図3)
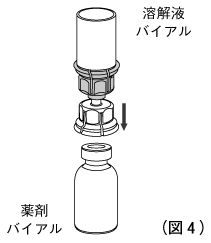
5. 薬剤バイアルを水平の台に置き、しっかりと握る。溶解器を付けた溶解液バイアルを逆さまにして、バイアル全体をしっかりと握り、溶解器の透明側アダプターの穿刺部を薬剤バイアルのゴム栓にまっすぐ下向きに刺し込む。このとき溶解液が薬剤バイアル中に移行します。(図4)
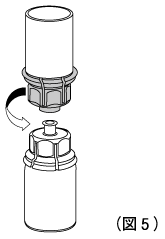
6. 片手で青色の部分をつかみ、もう片方の手で透明な部分をつかみ、慎重に回して二つに分ける。(図5)
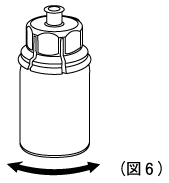
7. 透明な部分を付けたまま、薬剤バイアルを泡立てないように緩やかに揺り動かして完全に溶解する。バイアルを振らないこと。(図6)
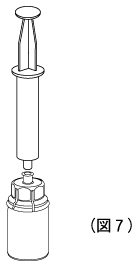
8. 空の滅菌済注射器に空気を吸い込む。薬剤バイアルが直立した状態で、注射器を溶解器のルアーロックに接続し、薬剤バイアルの中に空気を注入する。(図7)
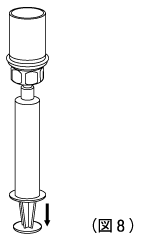
9. 注射器のプランジャーを押したまま、薬剤バイアルごと全体を上下逆さまにして、プランジャーをゆっくりと引っ張りながら、薬液を注射器の中に吸引する。(図8)
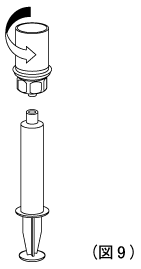
10. 薬液が注射器の中に移行したら、注射器のプランジャーを下向きにしたままの状態で、溶解器を注射器から取り外す。(図9)