1
社内資料:国内第I相試験(R2810-ONC-1622試験)[2022年12月23日承認、CTD2.7.2.2]
2
社内資料:国際共同第III相試験(R2810-ONC-1676試験)[2022年12月23日承認、CTD2.7.2.2]
3
社内資料:国際共同第III相試験(R2810-ONC-1676試験)[2022年12月23日承認、CTD2.7.3.2, CTD2.7.4.2]
4
社内資料:非臨床薬効薬理試験[2022年12月23日承認、CTD2.6.2.1]

抗悪性腫瘍剤ヒト型抗ヒトPD-1モノクローナル抗体
1瓶 450437円
有効成分 | セミプリマブ(遺伝子組換え)注) 350mg |
|---|---|
添加剤 | L-ヒスチジン 5.2mg L-ヒスチジン塩酸塩水和物 7.6mg 精製白糖 350mg L-プロリン 105mg ポリソルベート80 14mg |
| 剤形 | 注射剤(バイアル) |
|---|---|
| pH | 5.7~6.3 |
| 浸透圧比 | 約1.2(生理食塩液対比) |
| 性状 | 無色~微黄色で澄明又は乳白光を呈する液。半透明~白色の微粒子を認めることがある。 |
副作用 | 程度注) | 処置 |
間質性肺疾患 | Grade 2の場合 | Grade 1以下に回復するまで本剤を休薬する。 |
Grade 3以上又は再発性のGrade 2の場合 | 本剤を中止する。 | |
大腸炎・下痢 | Grade 2又は3の場合 | Grade 1以下に回復するまで本剤を休薬する。 |
Grade 4又は再発性のGrade 3の場合 | 本剤を中止する。 | |
肝機能障害 | ・AST又はALTが基準値上限の3~5倍まで増加した場合 ・総ビリルビンが基準値上限の1.5~3倍まで増加した場合 | Grade 1以下に回復するまで本剤を休薬する。 |
・AST又はALTが基準値上限の5倍超まで増加した場合 ・総ビリルビンが基準値上限の3倍超まで増加した場合 | 本剤を中止する。 | |
甲状腺機能低下症 甲状腺機能亢進症 甲状腺炎 | Grade 3以上の場合 | Grade 1以下に回復するまで本剤を休薬する。 |
副腎機能不全 | Grade 2以上の場合 | Grade 1以下に回復するまで本剤を休薬する。 |
下垂体炎 | Grade 2以上の場合 | Grade 1以下に回復するまで本剤を休薬する。 |
1型糖尿病 | Grade 3以上の場合 | Grade 1以下に回復するまで本剤を休薬する。 |
皮膚障害 | ・1週間以上続くGrade 2の場合 ・Grade 3の場合 ・Stevens-Johnson症候群(SJS)又は中毒性表皮壊死融解症(TEN)が疑われる場合 | Grade 1以下に回復するまで本剤を休薬する。 |
・Grade 4の場合 ・SJS又はTENが確認された場合 | 本剤を中止する。 | |
腎機能障害 | 血清クレアチニンが基準値上限又はベースラインの1.5~3倍まで増加した場合 | Grade 1以下に回復するまで本剤を休薬する。 |
血清クレアチニンが基準値上限又はベースラインの3倍超まで増加した場合 | 本剤を中止する。 | |
Infusion reaction | Grade 1又は2の場合 | 本剤の投与を中断又は投与速度を50%減速する。 |
Grade 3以上の場合 | 本剤を中止する。 | |
上記以外の副作用 | Grade 2又は3の場合 | Grade 1以下に回復するまで本剤を休薬する。12週間を超える休薬後もGrade 1以下まで回復しない場合には、本剤を中止する。 |
Grade 4又は再発性のGrade 3の場合 | 本剤を中止する。 |
10%以上 | 1~10%未満 | 1%未満 | 頻度不明 | |
|---|---|---|---|---|
感染症および寄生虫症 | 尿路感染 | 上気道感染 | ||
血液およびリンパ系障害 | 貧血 | 血小板減少症 | ||
免疫系障害 | シェーグレン症候群 | |||
神経系障害 | 頭痛 | |||
血管障害 | 高血圧 | |||
代謝および栄養障害 | 食欲減退 | |||
呼吸器、胸郭および縦隔障害 | 咳嗽、呼吸困難 | |||
胃腸障害 | 悪心、便秘、腹痛、嘔吐 | 口内炎 | ||
皮膚および皮下組織障害 | 発疹 | そう痒症 | ||
筋骨格系および結合組織障害 | 筋骨格痛 | 関節炎 | 筋力低下、リウマチ性多発筋痛 | |
腎および尿路障害 | 腎炎 | |||
一般・全身障害および投与部位の状態 | 疲労、発熱 | |||
臨床検査 | 血中クレアチニン増加、血中甲状腺刺激ホルモン増加 | 血中甲状腺刺激ホルモン減少 |
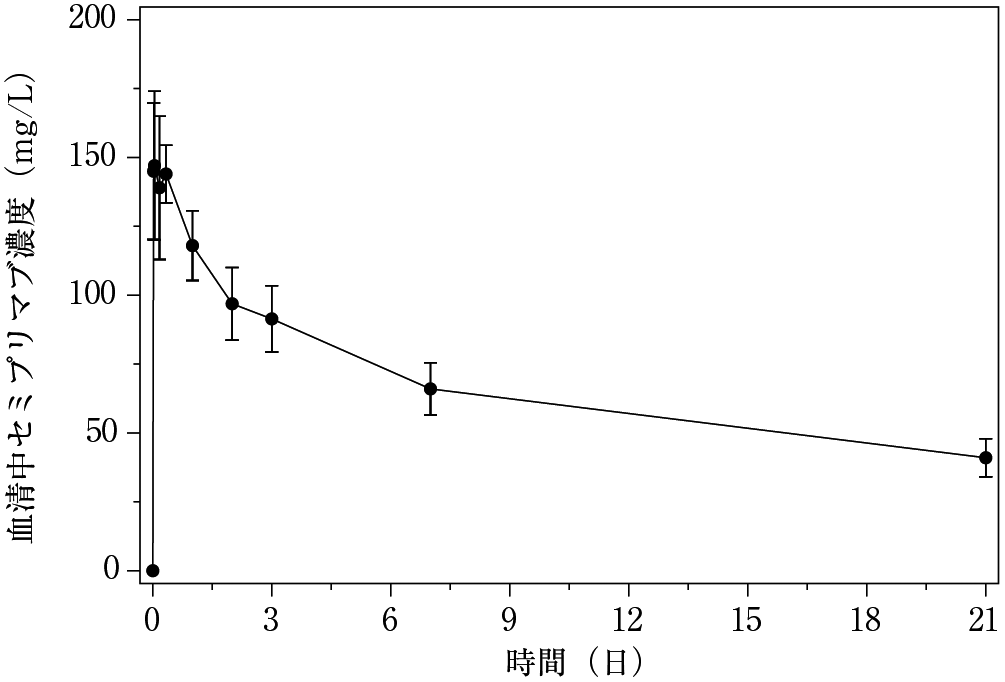
例数 | Ctrough(mg/L) | 例数 | Cmax(mg/L) | 例数 | AUC3w(mg・day/L) |
6 | 41.0±6.91 | 7 | 157±21.9 | 6 | 1345±99.7 |
初回投与後(サイクル1、1日目) | 定常状態(サイクル4、1日目) | ||||
例数 | Cmax(mg/L) | 例数 | Ctrough(mg/L) | 例数 | Cmax(mg/L) |
284 | 134±58.7 | 113 | 65.6±30.0 | 112 | 186±60.8 |
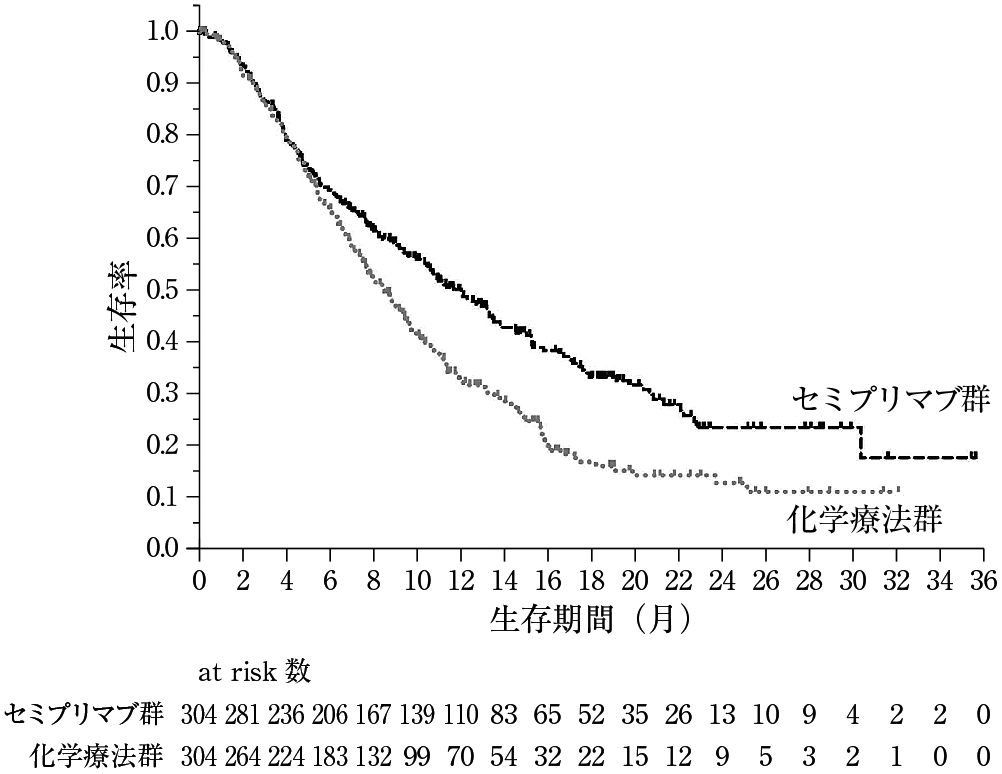
本剤群 (304例) | 化学療法群 (304例) | |
全生存期間注4) | ||
イベント数(%) | 184(60.5) | 211(69.4) |
中央値[月] (95%信頼区間) | 12.0(10.3, 13.5) | 8.5(7.5, 9.6) |
ハザード比注5) (95%信頼区間) | 0.685(0.560, 0.838) | |
p値注6) | 0.00011 | |