


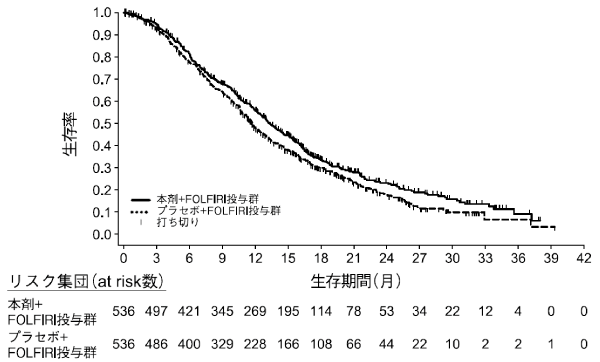
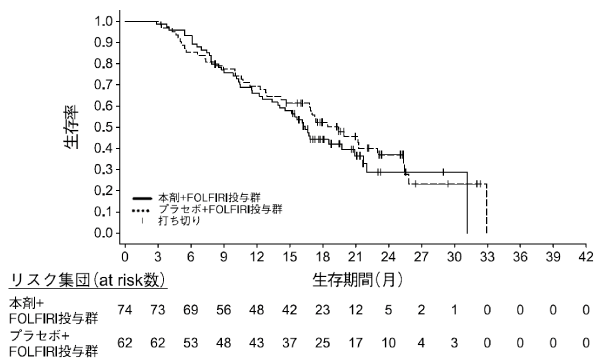
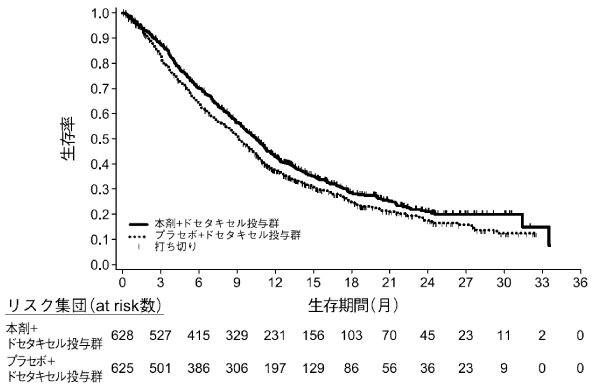
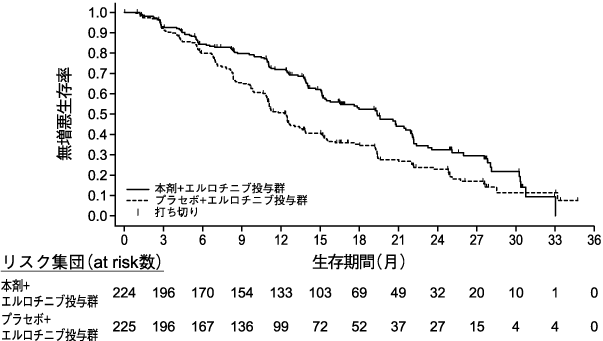
1
社内資料: ラムシルマブの生殖発生毒性に関する科学的評価(2015年3月26日承認、CTD2.6.6.6)
2
NDBを用いた調査結果の概要(VEGF/VEGFR阻害作用を有する薬剤の動脈解離に関するリスク評価): https://www.pmda.go.jp/files/000266521.pdf
3
社内資料: ラムシルマブの反復投与毒性試験(2015年3月26日承認、CTD2.6.6.3)
4
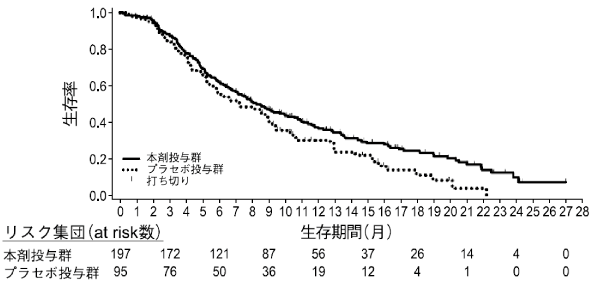
Yamaguchi K, et al.: Gastric Cancer. 2018; 21(6): 1041-1049
5
Ueda S, et al.: Oncologist. 2015; 20(5): 493-494
6
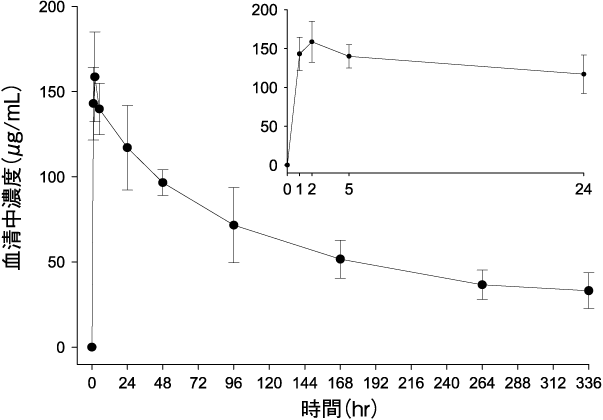
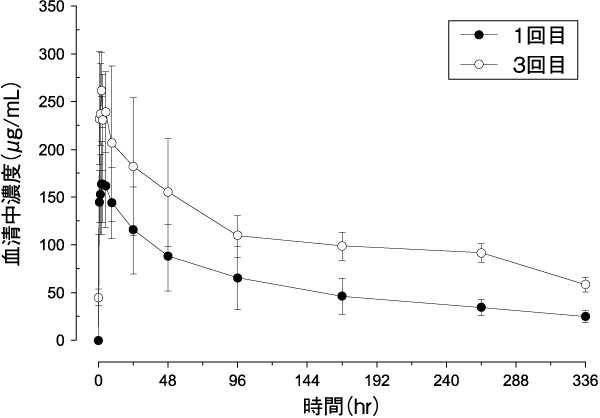
社内資料: 胃癌患者におけるラムシルマブ反復投与(パクリタキセル併用)後の薬物動態(第III相)(2015年3月26日承認、CTD2.7.6.12)
7
社内資料: 結腸・直腸癌患者におけるラムシルマブ反復投与(FOLFIRI併用)後の薬物動態(第III相)(2016年5月23日承認、CTD2.7.6.4)
8
社内資料: 日本人非小細胞肺癌患者におけるラムシルマブ反復投与(ドセタキセル併用)後の薬物動態(第II相)
9
Nakagawa K, et al.: Lancet Oncol. 2019; (doi: 10.1016/S1470-2045(19)30634-5)
10
社内資料: 非小細胞肺癌を対象とした国際共同第Ib/III相試験(RELAY試験 PartC)
11
社内資料: 肝細胞癌患者におけるラムシルマブ反復投与後の薬物動態(第III相)
12
Chow LQM, et al.: Cancer Chemother Pharmacol. 2016; 78(2): 433-441
13
Wang D, et al.: Cancer Chemother Pharmacol. 2016; 78(4): 727-733
14
Stein MN, et al.: Clin Pharmacol Biopharm. 2016; 5(4): 161-166
15
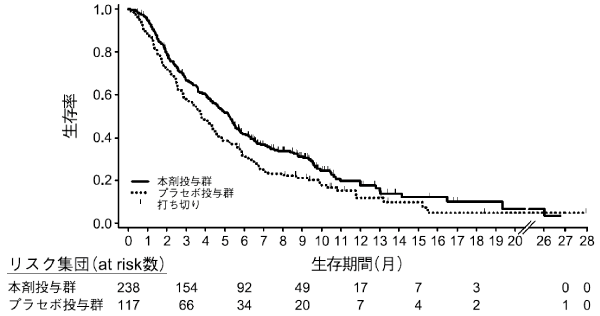
Fuchs CS, et al.: Lancet. 2014; 383(9911): 31-39
16
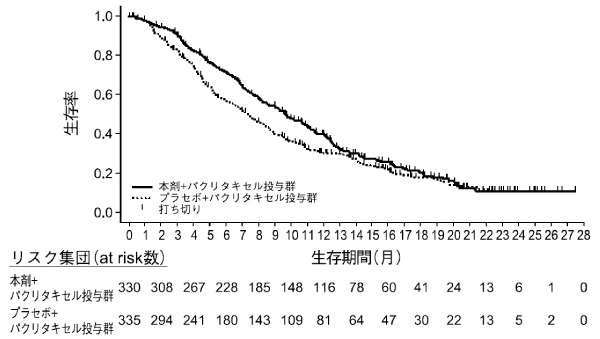
Wilke H, et al.: Lancet Oncol. 2014; 15(11): 1224-1235
17
Tabernero J, et al.: Lancet Oncol. 2015; 16(5): 499-508
18
Yoh K, et al.: Lung Cancer. 2016; 99: 186-193
19
Garon EB, et al.: Lancet. 2014; 384(9944): 665-673
20
Zhu AX, et al.: Lancet Oncol. 2019; 20(2): 282-296
21
社内資料: VEGFリガンドのVEGFR-2への結合に対するラムシルマブの作用(2015年3月26日承認、CTD2.6.2.2)
22
社内資料: ラムシルマブの細胞機能に対する作用(2015年3月26日承認、CTD2.6.2.2)
23
社内資料: ヒト胃癌のマウス異種移植モデルにおけるDC101の抗腫瘍効果(2015年3月26日承認、CTD2.6.2.2)
24
社内資料: ヒト結腸・直腸癌のマウス異種移植モデルにおけるDC101の抗腫瘍効果(2016年5月23日承認、CTD2.6.2.2)
25
社内資料: ヒト非小細胞肺癌のマウス異種移植モデルにおけるDC101の抗腫瘍効果
26
社内資料: ヒト肝細胞癌のマウス異種移植モデルにおけるDC101の抗腫瘍効果

