


1
国内のMDS患者にアザシチジンを皮下または静脈内投与した場合の薬物動態(ビダーザ注射用:2011年1月21日承認、申請資料概要2.7.1.2.2)
2
海外のMDS患者にアザシチジンを皮下または静脈内投与した場合の薬物動態(ビダーザ注射用:2011年1月21日承認、申請資料概要2.7.1.2.1)
3
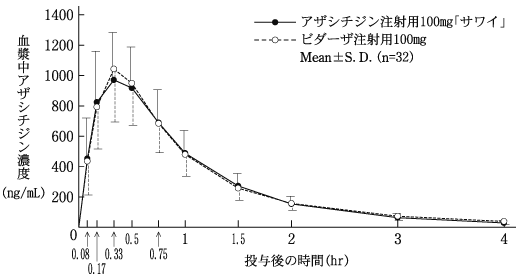
社内資料:生物学的同等性試験
4
in vitroヒト血清タンパク結合率(ビダーザ注射用:2011年1月21日承認、申請資料概要2.6.4.4.2)
5
in vitroヒト血球移行率(ビダーザ注射用:2011年1月21日承認、申請資料概要2.6.4.4.5)
6
Daher, G. C. et al.:Pharmacol. Ther., 1990;48:189-222
7
in vitro代謝試験①(ビダーザ注射用:2011年1月21日承認、申請資料概要2.6.4.5)
8
in vitro代謝試験②(ビダーザ注射用:2011年1月21日承認、審査報告書)
9
Troetel, W. M. et al.:Cancer Chemother. Rep., 1972;56:405-411
10
Israili, Z. H. et al.:Cancer Res., 1976;36:1453-1461
11
雄性ラットに静脈内投与又は皮下投与した後の尿及び糞中排泄率(ビダーザ注射用:2011年1月21日承認、申請資料概要2.6.4.6.3)
12
Laille, E. et al.:Pharmacotherapy, 2014;34:440-451
13
Uchida, T. et al.:Cancer Sci., 2011;102:1680-1686
14
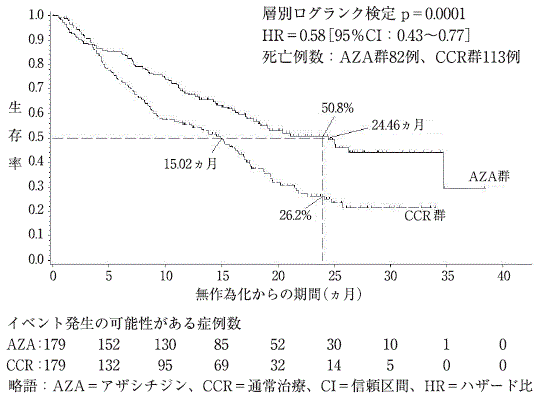
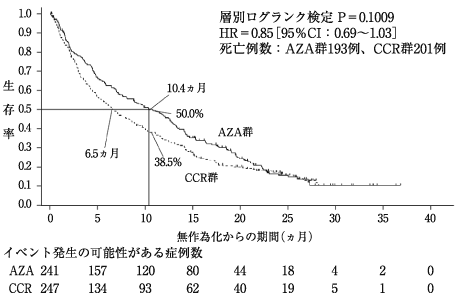
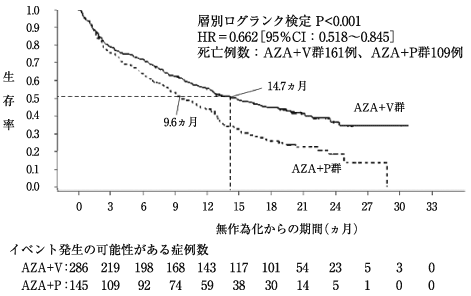
骨髄異形成症候群に対するアザシチジンの外国第Ⅲ相比較試験(AZA-001試験)(ビダーザ注射用:2011年1月21日承認、申請資料概要2.7.3.3.2.1)
15
Dombret, H. et al.:Blood, 2015;126:291-299
16
DiNardo, C. D. et al.:N. Engl. J. Med., 2020;383:617-629
17
Hollenbach, P. W. et al.:PLoS ONE, 2010;5:e9001
18
Hofmann, W. K. et al.:Leuk. Res., 2006;30:1347-1353
19
Khan, R. et al.:Exp. Hematol., 2006;34:35-43
20
Kimura, S. et al.:Anticancer Res., 2012;32:795-798

