1
Smeyne RJ, et al.: Nature. 1994; 368: 246-249
2
Klein R, et al.: Cell. 1993; 75: 113-122
3
Klein R, et al.: Nature. 1994; 368: 249-251


抗悪性腫瘍剤/トロポミオシン受容体キナーゼ阻害剤
1カプセル 4042.5円
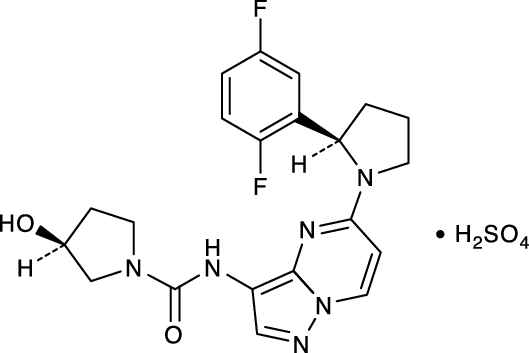
有効成分 | 1カプセル中ラロトレクチニブ 25mg含有 ラロトレクチニブ硫酸塩として 30.7mg |
|---|---|
添加剤 | カプセル内容物:なし カプセル本体中:ゼラチン、酸化チタン |
有効成分 | 1カプセル中ラロトレクチニブ 100mg含有 ラロトレクチニブ硫酸塩として 122.9mg |
|---|---|
添加剤 | カプセル内容物:なし カプセル本体中:ゼラチン、酸化チタン |
有効成分 | 1mL中ラロトレクチニブ 20.0mg含有 ラロトレクチニブ硫酸塩として 24.6mg |
|---|---|
添加剤 | ヒドロキシプロピル-β-シクロデキストリン、スクラロース、クエン酸ナトリウム水和物、無水クエン酸、安息香酸ナトリウム、香料 |
| 剤形 | 硬カプセル剤 |
|---|---|
| 色調 | 乳白色 |
| 外形 |  |
| 号数 | 2 |
| 識別コード |  |
| 剤形 | 硬カプセル剤 |
|---|---|
| 色調 | 乳白色 |
| 外形 |  |
| 号数 | 0 |
| 識別コード |  |
| 剤形 | 経口液剤 |
|---|---|
| 色・性状 | 無色~黄色、橙色、赤色又は帯褐色の液 |
用量調節段階 | 成人及び体表面積が1.0m2以上の小児の投与量 | 体表面積が1.0m2未満の小児の投与量 |
1段階減量 | 1回75mgを1日2回経口投与 | 1回75mg/m2(体表面積)を1日2回経口投与 |
2段階減量 | 1回50mgを1日2回経口投与 | 1回50mg/m2(体表面積)を1日2回経口投与 |
3段階減量 | 1回100mgを1日1回経口投与 | 1回25mg/m2(体表面積)を1日2回経口投与※2 |
程度 | 処置 |
グレード2 | 慎重に経過観察し、休薬・減量を考慮する。 |
グレード3又は4 | ベースライン又はグレード1以下に回復するまで休薬する。
|
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
強力な又は中程度のCYP3A阻害剤 イトラコナゾール、クラリスロマイシン、ボリコナゾール等 グレープフルーツ含有食品 | 本剤の副作用が増強されるおそれがあるので、これらの薬剤との併用は可能な限り避け、やむを得ず併用する場合には本剤の減量を考慮するとともに、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | これらの薬剤がCYP3Aを阻害することにより、本剤の血漿中濃度が上昇する可能性がある。 |
強力な又は中程度のCYP3A誘導剤 リファンピシン、フェニトイン、フェノバルビタール等 セイヨウオトギリソウ(セント・ジョーンズ・ワート)含有食品 | 本剤の有効性が減弱するおそれがあるので、これらの薬剤との併用は可能な限り避けること。 | これらの薬剤がCYP3Aを誘導することにより、本剤の血漿中濃度が低下する可能性がある。 |
CYP3Aの基質となる薬剤 シクロスポリン、キニジン、タクロリムス等 | これらの薬剤の副作用が増強されるおそれがあるので、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | 本剤がCYP3Aを阻害することにより、これらの薬剤の血漿中濃度が上昇する可能性がある。 |
5%以上 | 5%未満 | |
|---|---|---|
胃腸障害 | 悪心(10.6%)、便秘(10.1%)、味覚異常、嘔吐、下痢 | |
筋骨格系および結合組織障害 | 筋肉痛 | 筋力低下 |
一般・全身障害および投与部位の状態 | 疲労(14.3%)、浮腫 | |
神経系障害 | 頭痛 | |
皮膚および皮下組織障害 | 発疹 | |
その他 | 体重増加 | Al-P増加 |
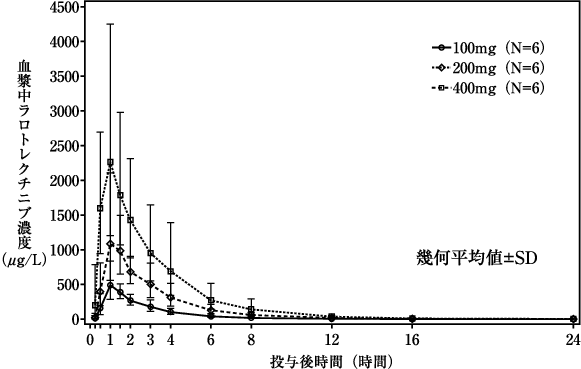
投与量 (mg) | n | Cmax (μg/L) | tmax※ (h) | AUC (μg・h/L) | t1/2 (h) |
100 | 6 | 548 (33.2) | 1.00 (1.0-3.0) | 1220 (23.9) | 1.88 (14.8) |
200 | 6 | 1250 (47.3) | 1.25 (1.0-3.0) | 3280 (33.8) | 2.55 (18.6) |
400 | 6 | 2730 (35.1) | 1.00 (0.5-4.0) | 7210 (42.2) | 2.78 (14.1) |
投与量 (mg) | 測定日 | n | Cmax (μg/L) | tmax※1 (h) | AUC12h (μg・h/L) | t1/2 (h) |
100 | 第1日目 | 39 | 868 (86.6) | 0.750 (0.250-2.05) | 2040 (92.6) | 1.68 (32.3) |
第8日目 | 37 | 788 (80.6) | 1.00 (0.500-9.37) | 2180 (97.2) | 2.73 (50.8)※2 | |
150 | 第1日目 | 7 | 923 (51.6) | 0.920 (0.530-1.00) | 2240 (47.0) | 1.55 (16.0) |
第8日目 | 6 | 815 (52.0) | 0.760 (0.500-2.00) | 2370 (59.6) | 2.16 (39.5)※3 | |
200 | 第1日目 | 6 | 1210 (122) | 0.760 (0.500-1.95) | 3760 (114)※4 | 1.67 (25.6)※4 |
第8日目 | 6 | 929 (175) | 1.03 (0.500-4.00) | 3040 (118) | 2.67 (49.3)※5 |
測定日 | n | Cmax (μg/L) | tmax※1 (h) | AUC12h (μg・h/L) | t1/2 (h) |
第1サイクル 第1日目 | 15 | 867 (51.1) | 1.00 (0.03-2.22) | 2220 (76.4) | 2.12 (35.9)※2 |
第4サイクル 第1日目 | 6 | 854 (43.9) | 0.500 (0.480-1.98) | 1470 (28.1) | 2.30 (15.4)※3 |
癌腫 | 奏効例数/評価可能例数 | 奏効率(%) (80%信頼区間) |
非小細胞肺癌 | 7/9 | 77.8 (51.0、93.9) |
甲状腺癌 | 13/19 | 68.4 (51.1、82.5) |
肉腫 | 15/19 | 78.9 (62.2、90.5) |
結腸直腸癌 | 3/8 | 37.5 (14.7、65.5) |
唾液腺癌 | 14/16 | 87.5 (70.0、96.6) |
胆道癌 | 0/2 | 0※1 |
中枢神経系原発腫瘍 | 1/7 | 14.3 (1.5、45.3)※2 |
固形癌 | 6/14 | 42.9 (24.3、63.1) |
癌腫 | 奏効例数/評価可能例数 | 奏効率(%) (95%信頼区間) |
乳児線維肉腫 | 22/22 | 100.0 (84.6、100.0) |
軟部肉腫 | 8/11 | 72.7 (39.0、94.0) |
中枢神経系原発腫瘍 | 3/8 | 37.5 (8.5、75.5)※1 |
骨肉腫 | 1/1 | 100.0※2 |
先天性間葉芽腎腫 | 1/1 | 100.0※2 |
悪性黒色腫 | 0/1 | 0※2 |