






1
社内資料:反復投与毒性試験(2019年9月20日承認,CTD 2.6.6.3)
2
社内資料:生殖発生毒性試験(2019年9月20日承認,CTD 2.6.6.6)
3
社内資料:分布・排泄に関する検討(2019年9月20日承認,CTD 2.6.4.4)
4
Seymour JF, et al.: N Engl J Med. 2018;378:1107-1120
5
社内資料:海外第Ⅰ相試験(M12-175試験)(2019年9月20日承認,CTD 2.7.2.2)
6
社内資料:国内第Ⅰ/Ⅱ相試験(M13-834試験)(2019年9月20日承認,CTD 2.7.2.2、2.7.3.1)
7
社内資料:海外第Ⅰ/Ⅱ相試験(M14-387試験)(2021年3月23日承認,CTD 2.7.2.2)
8
社内資料:食事の影響に関する試験(2019年9月20日承認,CTD 2.7.2.3)
9
社内資料:代謝に関する検討(2019年9月20日承認,CTD 2.6.4.5)
10
社内資料:マスバランス試験(2019年9月20日承認,CTD 2.7.2.2)
11
社内資料:肝機能障害患者における試験(2019年9月20日承認,CTD 2.7.2.2)
12
社内資料:ケトコナゾールとの薬物相互作用試験(2019年9月20日承認,CTD 2.7.2.2)
13
社内資料:リトナビルとの薬物相互作用試験(2019年9月20日承認,CTD 2.7.2.2)
14
社内資料:リファンピシンとの薬物相互作用試験(2019年9月20日承認,CTD 2.7.2.2)
15
社内資料:アジスロマイシンとの薬物相互作用試験(2019年9月20日承認,CTD 2.7.2.2)
16
社内資料:ワルファリンとの薬物相互作用試験(2019年9月20日承認,CTD 2.7.2.2)
17
社内資料:ジゴキシンとの薬物相互作用試験(2019年9月20日承認,CTD 2.7.2.2)
18
社内資料:海外第Ⅰb相(M14-358)試験(2021年3月23日承認,CTD 2.7.2.2)
19
社内資料:生理学的薬物動態モデルによるシミュレーション(2019年9月20日承認,CTD 2.7.2.3)
20
社内資料:トランスポーターを介した薬物相互作用試験(2019年9月20日承認,CTD 2.6.4.7)
21
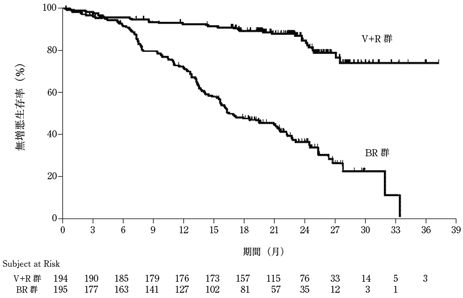
社内資料:海外第Ⅲ相試験(MURANO試験)(2019年9月20日承認,CTD 2.7.3.1)
22
DiNardo CD, et al.: N Engl J Med. 2020;383:617-629
23
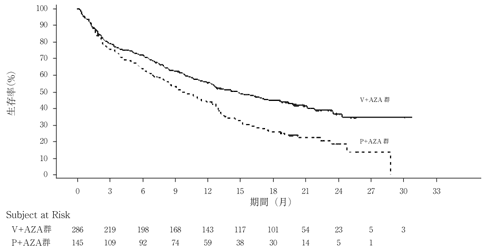
社内資料:海外第Ⅲ相試験(VIALE-A[M15-656]試験)(2021年3月23日承認,CTD 2.7.3)
24
Wei AH, et al.: Blood. 2020;135:2137-2145
25
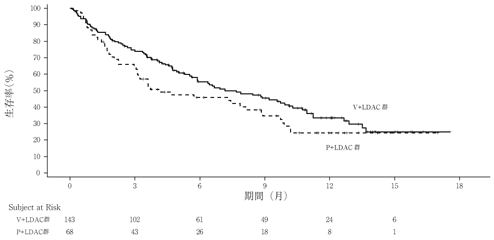
社内資料:海外第Ⅲ相試験(VIALE-C[M16-043]試験)(2021年3月23日承認,CTD 2.7.3)
26
社内資料:in vitro薬理試験(2019年9月20日承認,CTD 2.6.2.2)
27
社内資料:ex vivo薬理試験(2019年9月20日承認,CTD 2.6.2.2.3)

