







1
社内資料(遺伝毒性試験, 2017)(2018年1月19日承認、CTD2.6.6.4)
2
社内資料(日本人固形癌患者における薬物動態, 2014)(2018年1月19日承認、CTD2.7.6.2.4.2.4、2.7.2.3.1.6)
3
Ruth Plummer R, et al. Cancer Chemother Pharmacol. 2015;76(4):723-729.
4
社内資料(血漿蛋白結合[in vitro試験], 2017)(2018年1月19日承認、CTD2.7.2.3.1.2、2.7.2.4.2.1.1、2.7.2.4.2.1.2、2.7.2.4.2.1.3)
5
社内資料(代謝に関与する代謝酵素[in vitro試験], 2010)(2018年1月19日承認、CTD2.6.4.5.3)
6
社内資料(ヒトに[14C]-オラパリブを投与したマスバランス試験, 2009)(2018年1月19日承認、CTD 2.7.2.3.1.3、2.7.2.3.1.4、2.6.4.5.4.2.4、2.6.5.9.3)
7
社内資料(肝機能障害を有する固形癌患者における薬物動態, 2016)(2018年7月2日承認、CTD2.7.2.3.2.3)
8
社内資料(腎機能障害を有する固形癌患者における薬物動態, 2015)(2018年1月19日承認、CTD2.7.2.3.2.4)
9
Dirix L, et al. Clin Ther. 2016;38(10):2286-2299.
10
社内資料(CYPに対する阻害作用[in vitro試験], 2014)(2018年1月19日承認、CTD2.7.2.3.3.2.2、2.7.2.2.3.1)
11
社内資料(CYPに対する誘導作用[in vitro試験], 2015)(2018年1月19日承認、CTD2.7.2.3.3.2.2、2.7.2.2.3.1)
12
社内資料(UGTに対する阻害作用[in vitro試験], 2019)(2020年12月25日承認、CTD2.6.4.7.1、2.6.5.15.1)
13
社内資料(内分泌療法剤の相互作用, 2015)(2018年1月19日承認、CTD2.7.2.2.2.10、2.7.6.2.6.2.3)
14
社内資料(P-糖蛋白質の関与, 2007)(2018年1月19日承認、CTD2.7.2.4.2.4.1)
15
社内資料(トランスポーターに対する阻害作用, 2014)(2018年1月19日承認、CTD2.7.2.3.3.2.2)
16
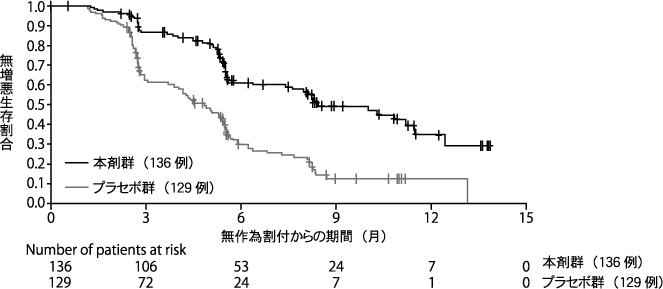
Pujade-Lauraine E, et al. Lancet Oncol. 2017;18:1274-1284.
17
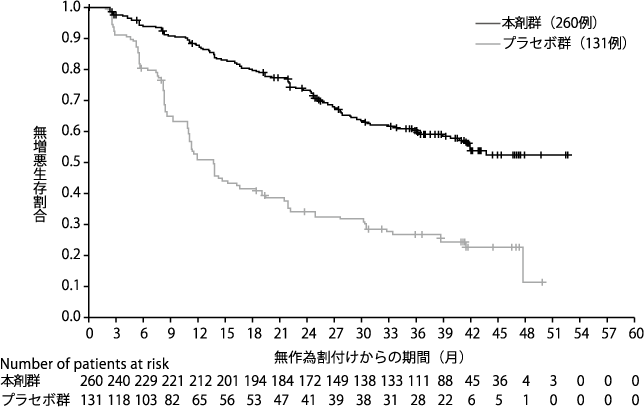
Ledermann J, et al. N Engl J Med. 2012;366:1382-1392.
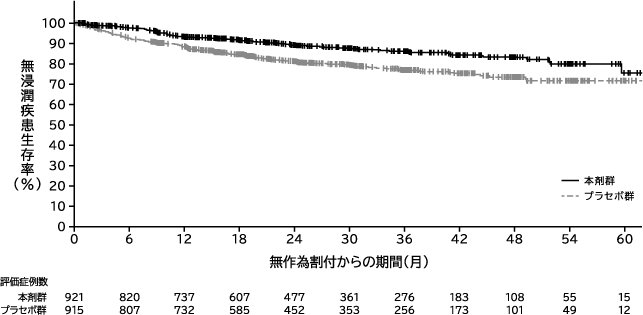
18
Moore K, et al. N Engl J Med. 2018;379:2495-2505.
19
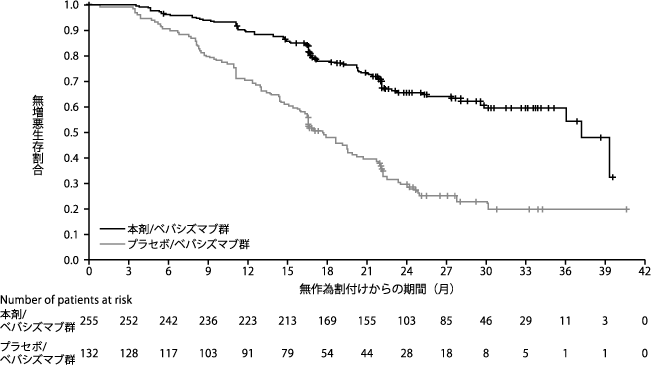
Ray-Coquard I, et al. N Engl J Med. 2019;381:2416-2428.
20
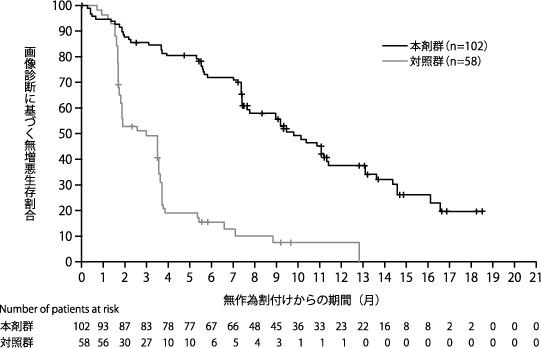
Robson M, et al. N Engl J Med. 2017;377:523-533.
21
Tutt ANJ, et al. N Engl J Med. 2021;384:2394-2405.
22
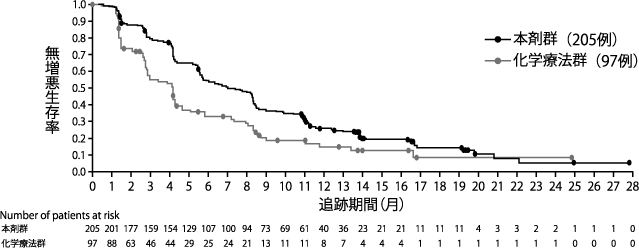
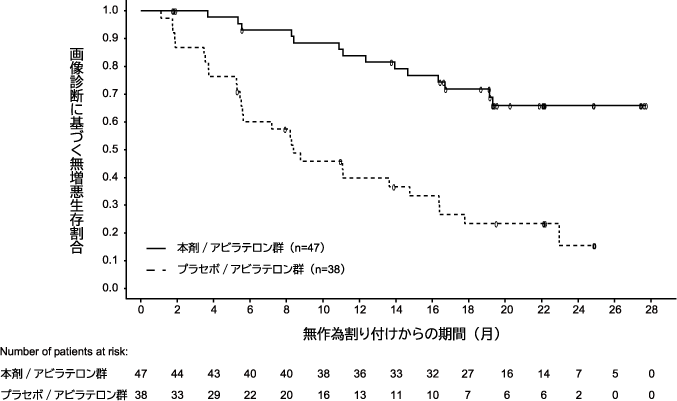
de Bono J, et al. N Engl J Med. 2020;382:2091-2102.
23
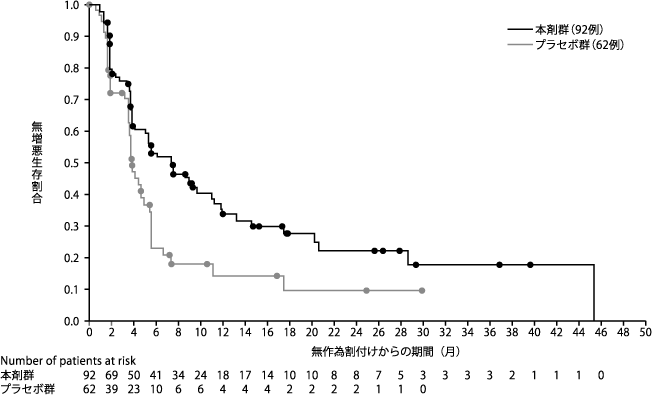
Clarke NW, et al. N Engl J Med Evid. 2022;1(9) doi: 10.1056/EVIDoa2200043
24
Golan T, et al. N Engl J Med. 2019;381:317-327.
25
Menear KA, et al. J Med Chem. 2008;51:6581-6591.
26
社内資料(各種腫瘍細胞株の増殖に対するオラパリブの作用[in vitro試験], 2013)(2018年1月19日承認、CTD2.6.2.2.3)
27
社内資料(HBCx-10腫瘍移植モデルにおけるオラパリブのPK、PD及び有効性の評価[in vivo試験], 2016)(2018年1月19日承認、CTD2.6.2.2.5)

