


1
Moltó A, et al.:Front. Med. 2018;62(5):1-10
2
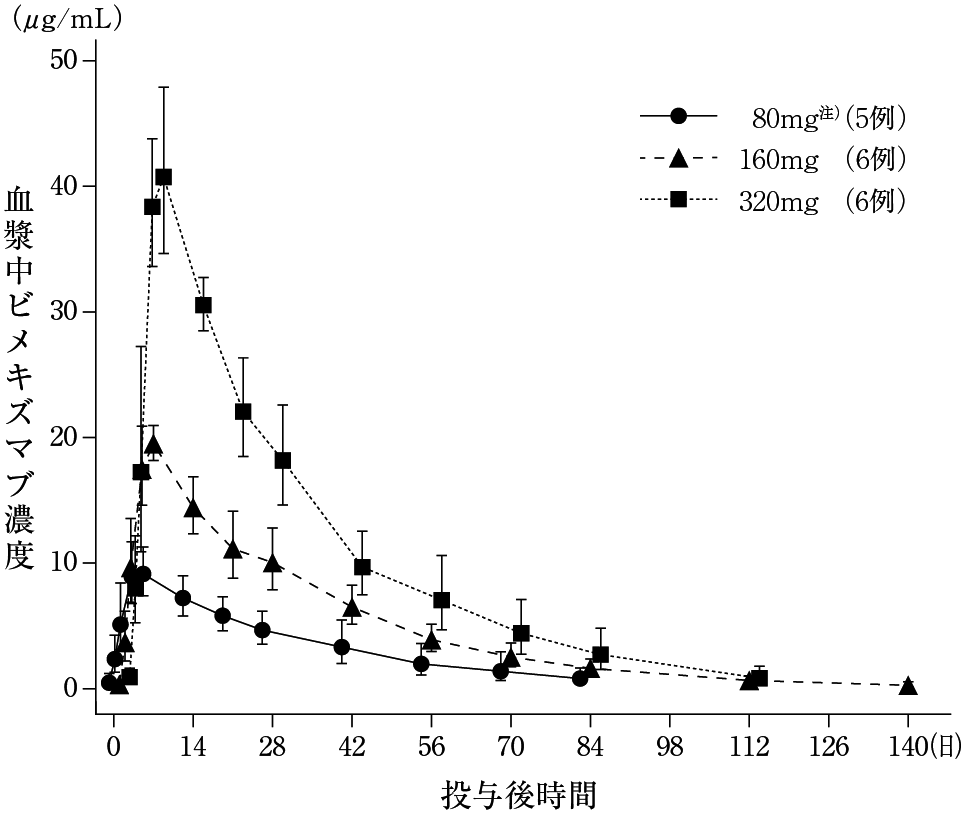
社内資料:臨床薬理試験成績 UP0042試験(2022年1月20日承認、CTD 2.7.6.3.1)
3
社内資料:国際共同第III相二重盲検比較試験成績 PS0009試験(2022年1月20日承認、CTD 2.7.6.6.5)
4
社内資料:国際共同第III相非盲検試験成績 PS0014試験(2022年1月20日承認、CTD 2.7.6.7.2)
5
社内資料:国際共同第III相二重盲検比較試験成績 PA0010試験
6
社内資料:国際共同第III相二重盲検比較試験成績 PA0011試験
7
社内資料:国際共同第III相二重盲検比較試験成績 AS0011試験
8
社内資料:国際共同第III相二重盲検比較試験成績 AS0010試験
9
社内資料:母集団薬物動態解析結果 CL0453(2022年1月20日承認、CTD 2.7.1.2.3)
10
社内資料:母集団薬物動態解析結果 CL0485(2022年1月20日承認、CTD 2.7.2.2.2.5)
11
社内資料:母集団薬物動態解析結果 CL0538
12
社内資料:カニクイザルを用いたePPND試験(2022年1月20日承認、CTD 2.6.6.6.1)
13
社内資料:国際共同第III相二重盲検比較試験成績 PS0008試験(2022年1月20日承認、CTD 2.7.6.6.4)
14
社内資料:国際共同第III相二重盲検比較試験成績 PS0013試験(2022年1月20日承認、CTD 2.7.6.6.6)
15
社内資料:ビメキズマブのIL-17A及びIL-17Fに対する選択的結合(2022年1月20日承認、CTD 2.6.2.2.1.3)
16
社内資料:IL-17AとIL-17Fの中和作用(2022年1月20日承認、CTD 2.6.2.2.1.4)
17
社内資料:In vitroにおける炎症性遺伝子発現に対する影響(2022年1月20日承認、CTD 2.6.2.2.1.1)
18
社内資料:In vitroにおける好中球及び単球の遊走に対する阻害作用(2022年1月20日承認、CTD 2.6.2.2.1.5)

