


1
Kimball, A.B., et al.:Br. J. Dermatol. 2015;173:1183-1190
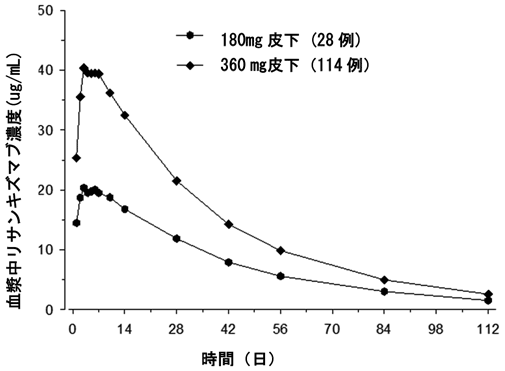
2
社内資料:健康成人における第Ⅰ相試験(2019年3月26日承認、CTD2.7.2.2)
3
社内資料:健康成人における第Ⅰ相試験(2022年9月26日承認、CTD2.7.2.2)
4
社内資料:健康成人における第Ⅰ相試験(2022年9月26日承認、CTD2.7.1.2)
5
社内資料:母集団薬物動態(2022年9月26日承認、CTD2.7.2.3)
6
社内資料:CYP基質薬物との相互作用試験(2019年3月26日承認、CTD 2.7.2.2.1)
7
社内資料:維持療法試験M16-000試験(2022年9月26日承認、CTD2.7.3.2、CTD2.7.3.3)
8
社内資料:IL-23に対する結合親和性(2019年3月26日承認、CTD2.6.2.2)
9
社内資料:in vitro中和作用(2019年3月26日承認、CTD2.6.2.1)
10
社内資料:in vivo中和作用(2019年3月26日承認、CTD2.6.2.2)

