


1
Osamu N, et al. : J Dermatol Sci. 2014; 75: 201-204
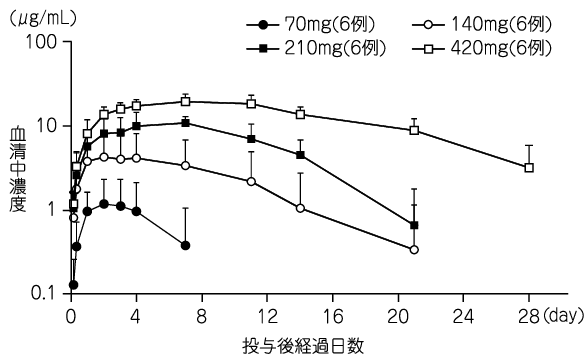
2
社内資料:局面型皮疹を有する乾癬患者を対象とした第Ⅱ相臨床試験(2016年7月4日承認、CTD2.7.6.7)
3
社内資料:体軸性脊椎関節炎患者を対象とした第Ⅲ相臨床試験における地域間の薬物動態の比較(2020年11月27日承認、CTD2.7.2.3.3)
4
社内資料:掌蹠膿疱症患者を対象とした第Ⅲ相プラセボ対照二重盲検比較及び非盲検継続投与試験(2023年8月23日承認、CTD2.7.2.2.1)
5
Timmermann S, et al.:Basic Clin Pharmacol Toxicol. 2019; 125: 16-25
6
社内資料:体軸性脊椎関節炎患者を対象とした母集団薬物動態解析(2020年11月27日承認、CTD2.7.2.3.2)
7
社内資料:母集団薬物動態解析による民族差の検討(2016年7月4日承認、CTD2.7.2.3.2)
8
社内資料:中等度~重度の尋常性乾癬患者を対象とした薬物相互作用試験(2016年7月4日承認、CTD2.7.6.6)
9
社内資料:局面型皮疹を有する乾癬患者を対象とした第Ⅲ相臨床試験(2016年7月4日承認、CTD2.7.6.11)
10
社内資料:乾癬性関節炎患者を対象とした第Ⅱ相臨床試験(2016年7月4日承認、CTD2.7.6.12)
11
社内資料:膿疱性乾癬患者及び乾癬性紅皮症患者を対象とした第Ⅲ相臨床試験(2016年7月4日承認、CTD2.7.6.15)
12
社内資料:体軸性脊椎関節炎患者を対象とした第Ⅲ相臨床試験(2020年11月27日承認、CTD2.7.6.1)
13
社内資料:掌蹠膿疱症患者を対象とした第Ⅲ相プラセボ対照二重盲検比較及び非盲検継続投与試験(2023年8月23日承認、CTD2.7.6.1)
14
社内資料:ヒトIL-17RA細胞外ドメインに対する結合親和性(2016年7月4日承認、CTD2.6.2.2.1)
15
社内資料:ヒトIL-17AとIL-17RAの結合に対する競合阻害作用(2016年7月4日承認、CTD2.6.2.2.2)
16
社内資料:ヒト全血中IL-17RA陽性白血球細胞に対する結合性(2016年7月4日承認、CTD2.6.2.2.3)
17
社内資料:ヒトIL-17A、IL-17F、IL-17A/F刺激依存的GROα産生に対する阻害作用(2016年7月4日承認、CTD2.6.2.2.4)
18
社内資料:ヒトIL-17C刺激依存的DEFB4 mRNA発現に対する阻害作用(2016年7月4日承認、CTD2.6.2.2.5)
19
社内資料:ヒトIL-25刺激依存的IL-5産生に対する阻害作用(2016年7月4日承認、CTD2.6.2.2.6)
20
社内資料:マウス乾癬モデルに対する作用(2016年7月4日承認、CTD2.6.2.2.9)
21
Russell CB, et al. : J Immunol. 2014; 192: 3828-3836
22
社内資料:マウス炎症性関節炎モデルに対する作用(2016年7月4日承認、CTD2.6.2.2.10)

