


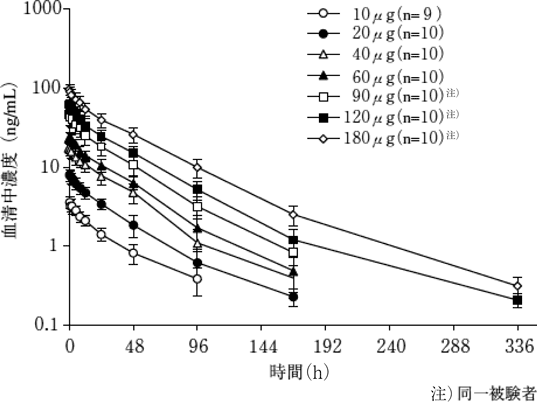
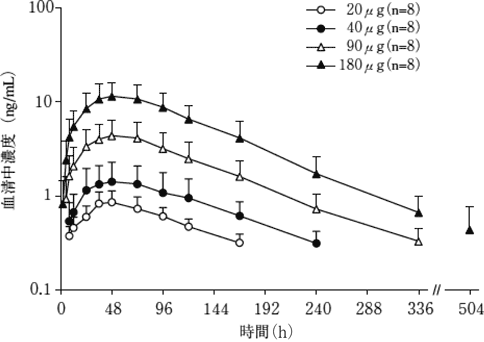
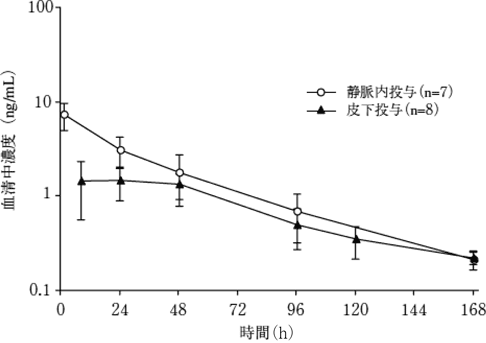
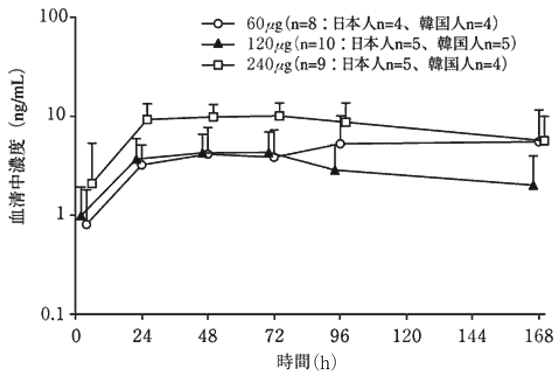
1
Hattori M, et al.:Clin Exp Nephrol. 2014; 18: 634-641
2
Besarab A, et al.:N Engl J Med. 1998; 339: 584-590
3
Singh AK, et al.:N Engl J Med. 2006; 355: 2085-2098
4
Pfeffer MA, et al.:N Engl J Med. 2009; 361: 2019-2032
5
Leyland-Jones B, et al.:J Clin Oncol. 2005; 23: 5960-5972
6
Henke M, et al.:Lancet. 2003; 362: 1255-1260
7
Overgaard J, et al.:J Clin Oncol. 2009; 27: 302s
8
Luksenburg H, et al.:FDA Briefing Document. ODAC May 4, 2004
9
Smith RE Jr, et al.:J Clin Oncol. 2008; 26: 1040-1050
10
菅朗ほか : 腎と透析. 2007; 63: 625-631
11
Uematsu T, et al.:Jpn J Clin Pharmacol Ther. 2007; 38: 331-339
12
飯野靖彦ほか:腎と透析. 2010; 68: 111-120
13
Uemura O, et al.:Clin Exp Nephrol. 2014; 18: 932-938
14
社内資料:骨髄異形成症候群患者を対象とした用量反応試験(2014年12月18日承認、CTD2.7.6.1)
15
社内資料:本剤反復投与による薬物動態の検討(2010年4月16日承認、CTD2.7.2.2)
16
社内資料:保存期慢性腎臓病患者における皮下投与時のバイオアベイラビリティ(2010年4月16日承認、CTD2.7.1.3)
17
社内資料:ラットにおける静脈内投与時の組織分布(2007年4月18日承認、CTD2.6.4.4)
18
社内資料:ラットにおける皮下投与時の組織分布(2010年4月16日承認、CTD2.6.4.4)
19
保利敬ほか : 腎と透析. 2007; 62: 679-691
20
Akizawa T, et al.:Ther Apher Dial. 2007; 11: 220-226
21
林晃正ほか : 腎と透析. 2010; 68: 931-945
22
Akizawa T, et al.:Ther Apher Dial. 2011; 15: 431-440
23
社内資料:腹膜透析患者を対象とした本剤の効果(第Ⅲ相)(2010年4月16日承認、CTD2.7.3.3.3.3)
24
永野伸郎ほか:腎と透析. 2006; 60: 1039-1046
25
社内資料:腎性貧血モデルラットにおける本剤及びエポエチン アルファ単回皮下投与時の貧血改善効果(2010年4月16日承認、CTD2.6.2.2)

