



 (刻印)
(刻印)
 (刻印)
(刻印)
 (刻印)
(刻印)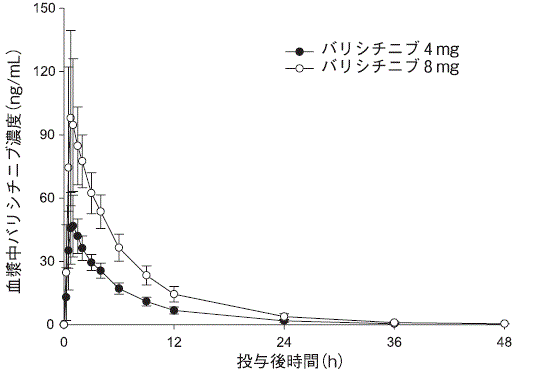
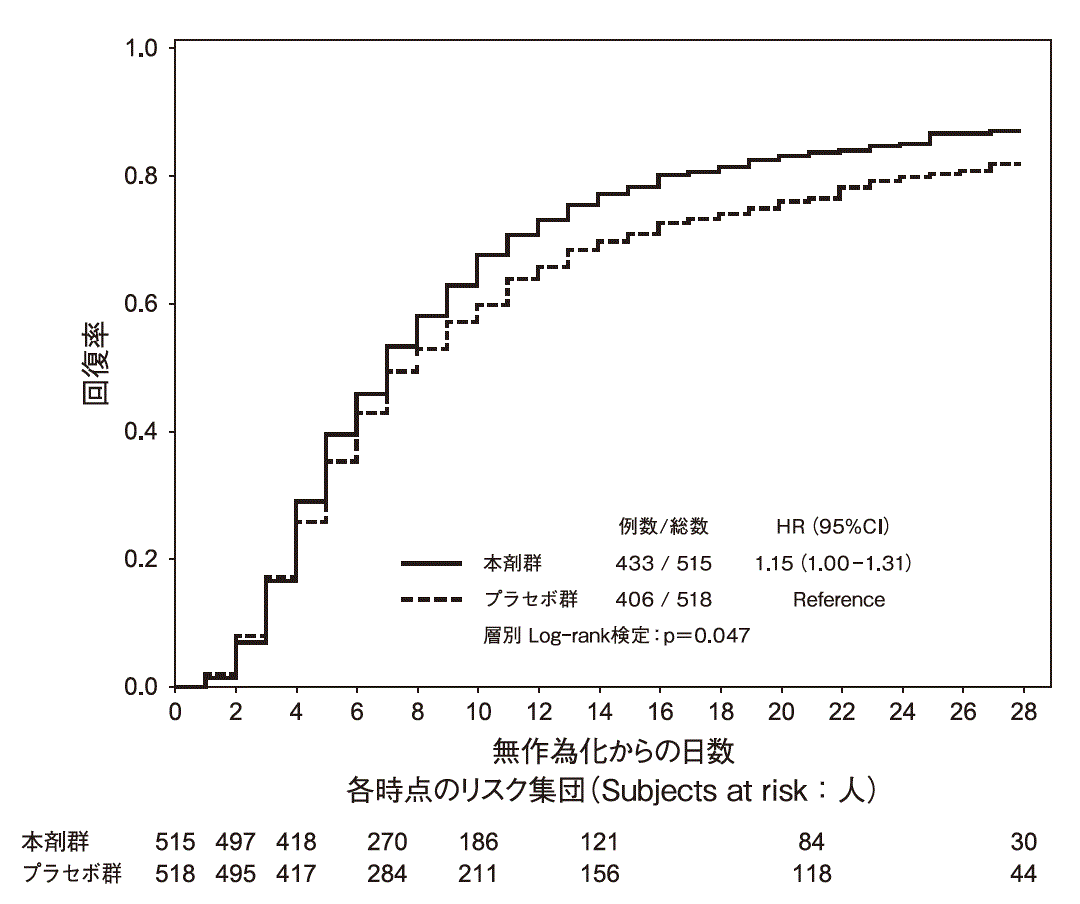
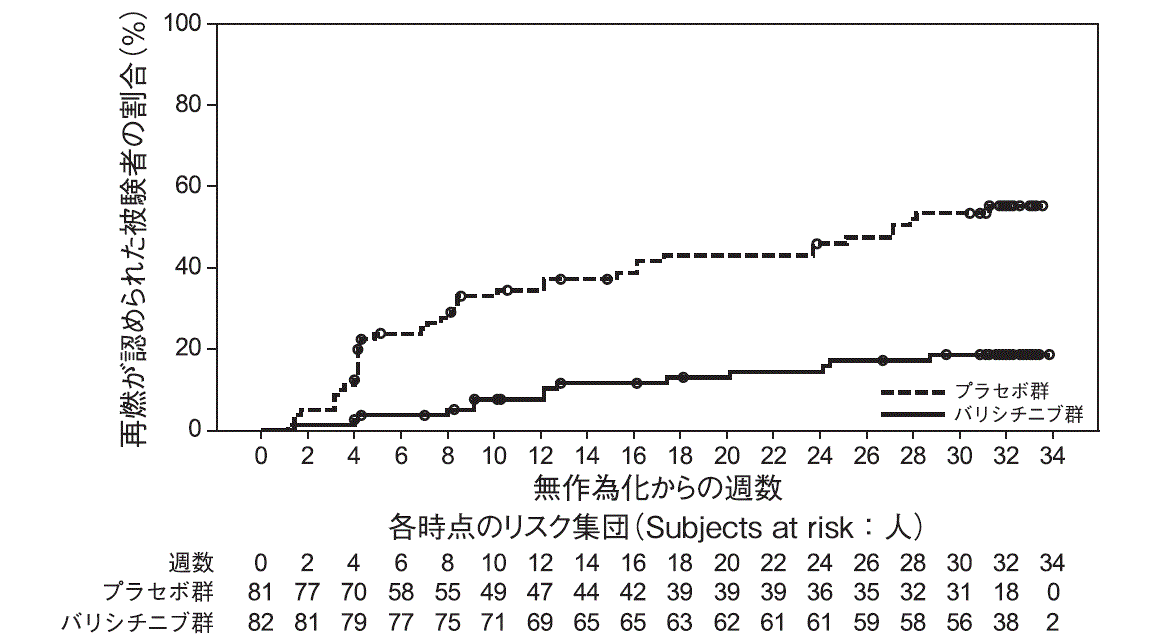
1
社内資料: バリシチニブの生殖発生毒性試験(2017年7月3日承認, CTD 2.6.6)
2
Smitten AL, et al.: Arthritis Res. Ther. 2008; 10(2): R45
3
社内資料: 日本人で相対的バイオアベイラビリティ及び食事の影響を評価した試験(2017年7月3日承認, CTD 2.7.2.2.2.1.2, 2.7.1.2.3.1)
4
社内資料: 関節リウマチ患者を対象とした母集団薬物動態解析(第II相試験及び第III相試験)(2017年7月3日承認, CTD 2.7.2.3.1.9.4.1)
5
社内資料: アトピー性皮膚炎患者を対象とした母集団薬物動態解析(第II相試験及び第III相試験)(2020年12月25日承認, CTD 2.7.2.3.1.5.1)
6
社内資料: アトピー性皮膚炎小児患者を対象とした母集団薬物動態解析(第III相試験)
7
社内資料: 円形脱毛症患者を対象とした母集団薬物動態解析(第II/III相試験)(2022年6月20日承認, CTD 2.7.2.1.2)
8
社内資料: 多関節に活動性を有する若年性特発性関節炎患者を対象とした母集団薬物動態解析(第III相試験)
9
社内資料: 絶対的バイオアベイラビリティを検討した試験(2017年7月3日承認, CTD 2.7.1.2.2.1)
10
社内資料: 外国人で相対的バイオアベイラビリティ及び食事の影響を評価した試験(2017年7月3日承認, CTD 2.7.1.2.3.2)
11
社内資料: 蛋白結合(in vitro; ラット、イヌ及びヒト血清及び血漿)(2017年7月3日承認, CTD 2.7.2.2.1.1)
12
社内資料: 代謝に関連するヒトCYP分子種の同定(in vitro)(2017年7月3日承認, CTD 2.7.2.2.1.3.1)
13
社内資料: 放射性標識体投与時の薬物動態試験(2017年7月3日承認, CTD 2.7.2.2.2.1.5, 2.7.2.3.1.6)
14
社内資料: 腎機能障害被験者における薬物動態試験(2017年7月3日承認, CTD 2.7.2.2.2.2.1)
15
社内資料: 肝機能障害被験者における薬物動態試験(2017年7月3日承認, CTD 2.7.2.2.2.2.2)
16
社内資料: P-gpの基質となる可能性及び阻害する可能性の評価(in vitro)(2017年7月3日承認, CTD 2.7.2.2.1.3.2)
17
社内資料: BCRPの基質となる可能性の評価(in vitro)(2017年7月3日承認, CTD 2.7.2.2.1.3.2, 2.7.2.2.1.4.3)
18
社内資料: OCT1、OCT2、OATP1B1、OAT1及びOAT3の基質となる可能性の評価(in vitro)(2017年7月3日承認, CTD 2.7.2.2.1.3.2)
19
社内資料: MATE1及びMATE2-Kの基質となる可能性及び阻害する可能性の評価(in vitro)(2017年7月3日承認, CTD 2.7.2.2.1.3.2)
20
社内資料: プロベネシドとの薬物相互作用試験(2017年7月3日承認, CTD 2.7.2.2.2.3.1.4)
21
社内資料: ケトコナゾール及びフルコナゾールとの薬物相互作用試験(2017年7月3日承認, CTD 2.7.2.2.2.3.1.1)
22
社内資料: リファンピシンとの薬物相互作用試験(2017年7月3日承認, CTD 2.7.2.2.2.3.1.2)
23
社内資料: シクロスポリンとの薬物相互作用試験(2017年7月3日承認, CTD 2.7.2.2.2.3.1.3)
24
社内資料: オメプラゾールとの薬物相互作用試験(2017年7月3日承認, CTD 2.7.2.2.2.3.1.6)
25
社内資料: メトトレキサートとの薬物相互作用試験(2017年7月3日承認, CTD 2.7.2.2.2.3.2.4, 2.7.2.2.2.3.1.7)
26
社内資料: CYPの阻害についての検討(in vitro)(2017年7月3日承認, CTD 2.7.2.2.1.4.1)
27
社内資料: CYPの誘導についての検討(in vitro)(2017年7月3日承認, CTD 2.7.2.2.1.4.2)
28
社内資料: OCT1、OCT2、OATP1B1、OAT1及びOAT3を阻害する可能性の評価(in vitro)(2017年7月3日承認, CTD 2.7.2.2.1.4.3)
29
社内資料: OAT2を阻害する可能性の評価(in vitro)
30
社内資料: OATP1B3の基質となる可能性及び阻害する可能性の評価(in vitro)(2017年7月3日承認, CTD 2.7.2.2.1.4.3)
31
社内資料: BCRPを阻害する可能性の評価(in vitro)(2017年7月3日承認, CTD 2.7.2.2.1.4.3)
32
社内資料: シンバスタチンとの薬物相互作用試験(2017年7月3日承認, CTD 2.7.2.2.2.3.2.1)
33
社内資料: 経口避妊薬との薬物相互作用試験(2017年7月3日承認, CTD 2.7.2.2.2.3.2.2)
34
社内資料: ジゴキシンとの薬物相互作用試験(2017年7月3日承認, CTD 2.7.2.2.2.3.2.3)
35
社内資料: MTXで効果不十分な関節リウマチ患者を対象とした第III相試験(RA-BEAM(JADV)試験)(2017年7月3日承認, CTD 2.7.6.3.6)
36
Taylor PC, et al.: N. Engl. J. Med. 2017; 376(7): 652-662
37
社内資料: 抗リウマチ薬の使用経験のない関節リウマチ患者を対象とした第III相試験(RA-BEGIN(JADZ)試験)(2017年7月3日承認, CTD 2.7.6.3.7)
38
Fleischmann R, et al.: Arthritis & Rheumatology. 2017; 69(3): 506-517
39
社内資料: MTXを含むcDMARDに対して効果不十分な関節リウマチ患者を対象とした第III相試験(RA-BUILD(JADX)試験)(2017年7月3日承認, CTD 2.7.6.3.5)
40
Dougados M, et al.: Ann. Rheum. Dis. 2017; 76(1): 88-95
41
社内資料: TNF阻害剤に対して効果不十分な関節リウマチ患者を対象とした第III相試験(RA-BEACON(JADW)試験)(2017年7月3日承認, CTD 2.7.6.3.4)
42
Genovese MC, et al.: N. Engl. J. Med. 2016; 374(13): 1243-1252
43
社内資料: 関節リウマチ患者を対象としたバリシチニブの長期安全性及び有効性を検討する多施設共同第III相試験(RA-BEYOND(JADY)試験)(2017年7月3日承認, CTD 2.7.6.3.8)
44
社内資料: 中等症から重症のアトピー性皮膚炎患者を対象に外用コルチコステロイドと併用した多施設共同無作為化二重盲検プラセボ対照第III相試験(BREEZE-AD7(JAIY)試験)(2020年12月25日承認, CTD 2.7.6.5)
45
社内資料: 中等症から重症のアトピー性皮膚炎患者を対象とした多施設共同無作為化二重盲検プラセボ対照第III相試験(BREEZE-AD1(JAHL)試験)(2020年12月25日承認, CTD 2.7.6.3)
46
社内資料: 中等症から重症のアトピー性皮膚炎患者を対象とした多施設共同無作為化二重盲検プラセボ対照第III相試験(BREEZE-AD2(JAHM)試験)(2020年12月25日承認, CTD 2.7.6.4)
47
社内資料: 中等症から重症のアトピー性皮膚炎小児患者を対象とした多施設共同無作為化二重盲検プラセボ対照第III相試験(BREEZE-AD-PEDS(JAIP)試験)
48
社内資料: NIAID ACTT-2試験(2021年4月23日承認, CTD 2.5.4, 2.5.5)
49
Kalil AC, et al.: N. Engl. J. Med. 2021; 384(9): 795-807
50
社内資料: 重症又は極めて重症の円形脱毛症を有する成人患者を対象とした多施設共同無作為化二重盲検プラセボ対照第II/III相試験(BRAVE-AA1(JAHO)試験)(2022年6月20日承認, CTD 2.7.6.2)
51
King B, et al.: N. Engl. J. Med. 2022; 386(18): 1687-1699
52
社内資料: 重症又は極めて重症の円形脱毛症を有する成人患者を対象とした多施設共同無作為化二重盲検プラセボ対照第III相試験(BRAVE-AA2(JAIR)試験)(2022年6月20日承認, CTD 2.7.6.3)
53
社内資料: 多関節に活動性を有する若年性特発性関節炎患者を対象とした多施設共同無作為化二重盲検プラセボ対照第III相試験(JUVE-BASIS(JAHV)試験)
54
Higashi Y,: Folia Pharmacol. Jpn. 2014; 144(4): 160-166
55
Fridman JS, et al.: J. Immunol. 2010; 184(9): 5298-5307
56
Shi JG, et al.: J. Clin. Pharmacol. 2014; 54(12): 1354-1361

