






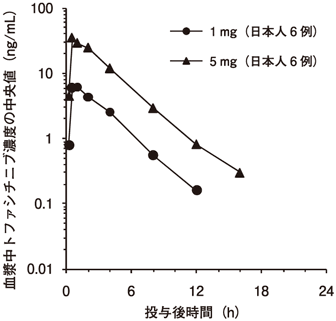
1
社内資料:生殖発生毒性試験(2013年3月25日承認 CTD2.6.6.6、2.6.6.9、2018年5月25日承認 CTD2.4.5) [L20120705046]
2
社内資料:授乳ラットにおける組織分布(2013年3月25日承認 CTD2.6.4.6、2.6.5.7) [L20120705038]
3
社内資料:外国第Ⅱ相試験 インフルエンザワクチン及び肺炎球菌ワクチン接種後の免疫応答への影響 [L20140701001]
4
社内資料:外国第Ⅱ/Ⅲ相試験 インフルエンザワクチン及び肺炎球菌ワクチン接種後の免疫応答への影響 [L20140701002]
5
社内資料:がん原性試験(2013年3月25日承認 CTD2.6.6.5、2.6.6.9) [L20120705045]
6
社内資料:単回及び反復投与毒性試験(サル)(2013年3月25日承認 CTD2.6.6.1、2.6.6.2、2.6.6.3) [L20120705041]
7
Borie, D. C. et al.:Transplantation. 2005;80(12):1756-1764
8
社内資料:健康成人における薬物動態(単回及び反復投与)(2013年3月13日承認 CTD2.7.2.2、2.7.2.3) [L20120705067]
9
社内資料:関節リウマチ患者におけるポピュレーションPK解析(2013年3月13日承認 CTD2.7.2.1、2.7.2.3) [L20120705091]
10
社内資料:潰瘍性大腸炎患者におけるポピュレーションPK解析(2018年5月25日承認 CTD2.7.2.3) [L20180327010]
11
社内資料:健康成人におけるバイオアベイラビリティの検討(2013年3月13日承認 CTD2.7.2.1) [L20120705064]
12
社内資料:健康成人における食事の影響(2013年3月13日承認 CTD2.7.2.2、2.7.2.3) [L20120705065]
13
社内資料:in vitroでの血漿蛋白結合率の検討 (2013年3月13日承認 CTD2.7.2.2、2.7.2.3) [L20120705092]
14
社内資料:in vitroでの各種蛋白との結合率の検討(2013年3月13日承認 CTD2.7.2.2、2.7.2.3) [L20120705093]
15
社内資料:血球への移行の検討(2013年3月13日承認 CTD2.7.2.2) [L20120705094]
16
社内資料:健康成人におけるマスバランスの検討 (2013年3月13日承認 CTD2.7.2.2、2.7.2.3) [L20120705066]
17
社内資料:in vitroでの代謝の検討(2013年3月13日承認 CTD2.7.2.2、2.7.2.3) [L20120705095]
18
社内資料:代謝に関連するヒトCYP酵素の検討 (2013年3月13日承認 CTD2.7.2.2、2.7.2.3) [L20120705096]
19
社内資料:ヒトの薬物代謝酵素に対する影響 (2013年3月13日承認 CTD2.7.2) [L20120705097]
20
Krishnaswami, S. et al.:J Clin Pharmacol. 2014;54(1):46-52
21
Lawendy, N. et al.:Clin Pharmacol Drug Dev. 2014;3(6): 421-427
22
社内資料:メトトレキサートとの薬物相互作用 (2013年3月13日承認 CTD2.7.2.2、2.7.2.3) [L20120705068]
23
Gupta, P. et al.:Clin Pharmacol Drug Dev. 2014;3(1): 72-77
24
社内資料:タクロリムス及びシクロスポリンとの薬物相互作用(2013年3月13日承認 CTD2.7.2.2、2.7.2.3) [L20120705072]
25
社内資料:リファンピシンとの薬物相互作用 (2013年3月13日承認 CTD2.7.2.2、2.7.2.3) [L20120705074]
26
Gupta, P. et al.:Br J Clin Pharmacol. 2012;74(1): 109-115
27
Menon, S. et al.:Clin Pharmacol Drug Dev. 2016;5(5):336-342
28
Klamerus, K. J. et al.:Clin Pharmacol Drug Dev. 2014;3(6): 499-507
29
社内資料:P糖蛋白質の基質としての評価試験 (2013年3月13日承認 CTD2.7.2.2、2.7.2.3) [L20120705098]
30
社内資料:P糖蛋白質阻害作用の検討(2013年3月13日承認 CTD2.7.2) [L20120705099]
31
社内資料:ヒト有機カチオントランスポーター(hOCT2)阻害作用の検討(2013年3月13日承認 CTD2.7.2) [L20120705100]
32
社内資料:健康成人における糸球体ろ過への影響の検討(2013年3月13日承認 CTD2.7.2.2、2.7.2.3、2.7.2.5) [L20120705077]
33
社内資料:ヒト有機カチオントランスポーター(hOCT1)の基質としての評価試験(2013年3月25日承認 CTD2.6.4.7、2018年5月25日承認 CTD2.6.4.7.3) [L20180327006]
34
社内資料:ヒト有機カチオントランスポーター(hOCT2)の基質としての評価試験(2013年3月25日承認 CTD2.6.4.7、2018年5月25日承認 CTD2.6.4.7.3) [L20180327007]
35
社内資料:ヒト有機アニオン輸送ポリペプチド(hOATP 1B1)阻害作用の検討(2013年3月13日承認 CTD2.7.2) [L20120705101]
36
社内資料:ヒト有機アニオン輸送ポリペプチド(hOATP 1B3)阻害作用の検討(2013年3月13日承認 CTD2.7.2) [L20120705102]
37
Giacomini, K. M. et al.:Nat Rev Drug Discov. 2010;9(3): 215-236
38
社内資料:ヒト有機アニオン輸送ポリペプチド(hOATP1B1及びhOATP1B3)の基質としての評価試験(2013年3月25日承認 CTD2.6.4.7、2018年5月25日承認 CTD2.6.4.7.3) [L20180327008]
39
社内資料:サンドイッチ培養ヒト肝細胞への取り込みの検討(2013年3月25日承認 CTD2.6.4.7、2018年5月25日承認 CTD2.4.3.6.3、2.6.4.7.3、2.6.5.15) [L20180327009]
40
Tanaka, Y. et al.:Arthritis Care Res. 2011;63(8):1150
41
Tanaka, Y. et al.:Mod Rheumatol. 2015;25(4): 514-521
42
Fleischmann, R. et al.:Arthritis Rheum. 2012 ; 64(3):617-629
43
Burmester, G. R. et al.:Lancet. 2013;381(9865):451-460
44
Fleischmann, R. et al.:N Engl J Med. 2012;367(6):495-507
45
社内資料:外国第Ⅲ相試験(DMARD効果不十分例、DMARD併用)(2013年3月25日承認 CTD2.7.6.26) [L20120705081]
46
van Vollenhoven, R. F. et al.:N Engl J Med. 2012;367(6):508-519
47
van der Heijde, D. et al.:Arthritis Rheum. 2013;65(3):559-570
48
社内資料:第Ⅲ相国際共同寛解導入試験(A3921094試験)(2018年5月25日承認 CTD2.7.6.2) [L20180327027]
49
社内資料:第Ⅲ相国際共同寛解維持試験(A3921096試験)(2018年5月25日承認 CTD2.7.6.4) [L20180327029]
50
ClinicalTrials.gov:Safety Study Of Tofacitinib Versus Tumor Necrosis Factor (TNF) Inhibitor In Subjects With Rheumatoid Arthritis (https://clinicaltrials.gov/ct2/home)
51
EU Clinical Trials Register:Phase 3b/4 Randomized Safety Endpoint Study of 2 Doses of Tofacitinib in Comparison to a Tumor Necrosis Factor (TNF) Inhibitor in Subjects with Rheumatoid Arthritis(https://www.clinicaltrialsregister.eu/ctr-search/search)

