










1
社内資料:生殖発生毒性試験(2020年1月23日承認,CTD 2.6.6.6)
2
社内資料:妊娠動物の組織分布(2020年1月23日承認,CTD 2.6.4.4)
3
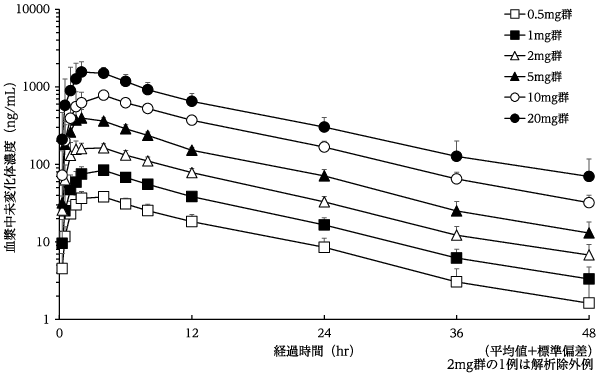
社内資料:健康成人における単回投与試験(2020年1月23日承認,CTD 2.7.6.2)
4
社内資料:健康成人における反復投与試験(2020年1月23日承認,CTD 2.7.6.4)
5
社内資料:健康成人における食事の影響試験(2020年1月23日承認,CTD 2.7.6.1)
6
社内資料:マスバランス試験(2020年1月23日承認,CTD 2.7.6.5)
7
社内資料:血漿蛋白結合率及び血球移行性(2020年1月23日承認,CTD 2.7.2.2)
8
社内資料:UGT及びSULT分子種による代謝試験(2020年1月23日承認,CTD 2.7.2.2)
9
社内資料:CYP阻害及び誘導作用(2020年1月23日承認,CTD 2.7.2.2)
10
社内資料:UGT阻害作用(2020年1月23日承認,CTD 2.7.2.2)
11
社内資料:薬物トランスポーター阻害作用(2020年1月23日承認,CTD 2.7.2.2)
12
Taniguchi T, et al.:J Pharmacol Exp Ther. 2019; 371(1):162-170
13
社内資料:腎機能障害者における臨床薬理試験(2020年1月23日承認,CTD 2.7.6.6)
14
Fukase H, et al.:Clin Exp Nephrol. 2020;24:S17-24
15
社内資料:肝機能障害者における臨床薬理試験(2020年1月23日承認,CTD 2.7.6.7)
16
Kumagai Y, et al.:Clin Exp Nephrol. 2020;24:S25-35
17
社内資料:高齢者男女における臨床薬理試験(2020年1月23日承認,CTD 2.7.6.8)
18
Nakatani H, et al.:Clin Exp Nephrol. 2020;24:S8-16
19
社内資料:健康成人における薬物相互作用試験(2020年1月23日承認,CTD 2.7.6.9)
20
Furihata K, et al.:Clin Exp Nephrol. 2020;24:S36-43
21
社内資料:第Ⅲ相ベンズブロマロン対照試験(2020年1月23日承認,CTD 2.7.6.15)
22
Hosoya T, et al.:Clin Exp Nephrol. 2020;24:S62-70
23
社内資料:第Ⅲ相フェブキソスタット対照試験(2020年1月23日承認,CTD 2.7.6.16)
24
Hosoya T, et al.:Clin Exp Nephrol. 2020;24:S71-79
25
社内資料:第Ⅲ相長期投与試験(2020年1月23日承認,CTD 2.7.6.17)
26
Hosoya T, et al.:Clin Exp Nephrol. 2020;24:S80-91
27
社内資料:ヒトURAT1発現細胞を用いた尿酸取り込み阻害試験(2020年1月23日承認,CTD 2.6.2.2)
28
社内資料:ヒトABCG2、OAT1及びOAT3発現細胞を用いた尿酸取り込み阻害試験(2020年1月23日承認,CTD 2.6.2.2)
29
社内資料:フサオマキザルを用いた血漿中尿酸値低下作用(2020年1月23日承認,CTD 2.6.2.2)

