



1
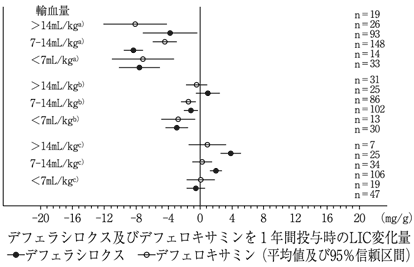
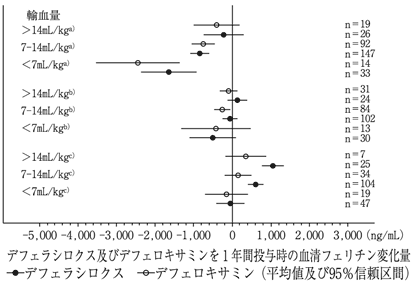
三谷絹子, et al.:厚生労働科学研究費補助金難治性疾患等政策研究事業 特発性造血障害に関する調査研究(令和1年度):輸血後鉄過剰症診療の参照ガイド2020:1-52 [20210078]
2
Takatoku,M.et al.:Eur.J.Haematol. 2007; 78(6): 487-494 [20072718]
3
Malcovati,L.et al.:J.Clin.Oncol. 2005; 23(30): 7594-7603 [20080836]
4
Olivieri,N.F.et al.:New Engl.J.Med. 1994; 331(9): 574-578 [19941365]
5
社内資料:国内第Ⅰ相試験(懸濁用錠)(2008年4月16日承認、CTD2.7.6.2.2.1) [20081244]
6
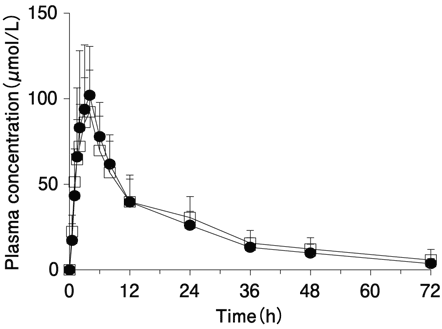
社内資料:懸濁用錠と顆粒の生物学的同等性(健康成人)(2017年7月3日承認、CTD2.7.6.1.2.4) [20170106]
7
社内資料:顆粒での食事の影響(2017年7月3日承認、CTD2.7.6.1.1.1) [20170107]
8
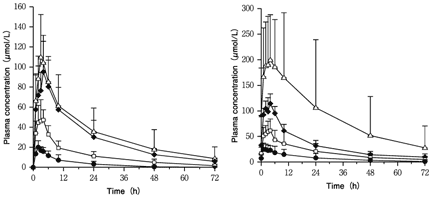
社内資料:外国健康成人血漿中濃度推移(懸濁用錠)(2008年4月16日承認、CTD2.7.2.3.1) [20081245]
9
社内資料:血漿蛋白質との結合(蛋白結合率)(2008年4月16日承認、CTD2.7.2.3.5) [20081247]
10
社内資料:結合蛋白の同定 (2008年4月16日承認、CTD2.7.2.3.5) [20081248]
11
社内資料:UDP-グルクロン酸転移酵素による抱合代謝 (2008年4月16日承認、CTD2.4.4.4.1) [20081249]
12
社内資料:チトクロムP450による代謝(2008年4月16日承認、CTD2.4.4.4.1) [20081250]
13
社内資料:経口投与時の吸収、血中動態、代謝及び排泄経路の検討を目的とした試験(懸濁用錠)(2008年4月16日承認、CTD2.7.2.3.7) [20081251]
14
社内資料:デフェラシロクスの薬物動態に対する肝機能障害の影響(懸濁用錠) [20120551]
15
Cappellini,M.D.et al.:Blood 2006; 107(9):3455-3462 [20076349]
16
Porter,J.et al.:Eur.J.Haematol. 2008; 80(2): 168-176 [20080837]
17
社内資料:海外第Ⅱ相及び第Ⅲ相試験(4試験併合)(懸濁用錠)(2008年4月16日承認、CTD2.7.3.3.2.5.2) [20081252]
18
Heinz,U.et al.:Angew. Chem. Int. Ed.1999; 38(17):2568-2570 [20080838]
19
Hershko,C.et al.:Blood 2001; 97(4): 1115-1122 [20080839]
20
社内資料:鉄負荷ラットにおける肝臓鉄への影響(2008年4月16日承認、CTD2.6.2.2.3.1.4) [20081253]
21
社内資料:非鉄負荷マーモセットにおける肝臓鉄への影響(2008年4月16日承認、CTD2.6.2.2.3.3.2) [20081254]

