




1
Kuchel O. et al.:J. Lab. Clin. Med., 1990;115:449-453
2
Engelman K. et al.:Circulation Research, 1966;18:I-104-I-109
3
Pyörälä K. et al.:Ann. Med. Int. fenn., 1968;57:65-73
4
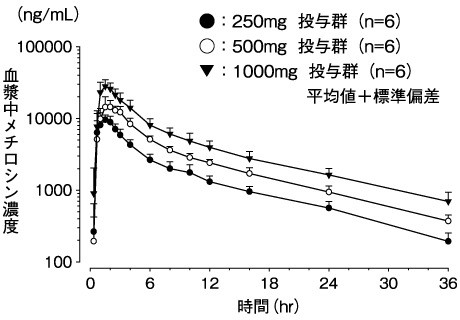
小野薬品工業:国内第Ⅰ相試験成績(社内資料;2019年1月8日承認、CTD2.7.6.1)
5
小野薬品工業:海外第Ⅰ相試験成績(社内資料;2019年1月8日承認、CTD2.7.6.2)
6
小野薬品工業:国内第Ⅰ/Ⅱ相試験成績(社内資料;2019年1月8日承認、CTD2.7.6.3)
7
Engelman K. et al. : J. Clin. Invest., 1968;47:568-576
8
Naruse M. et al.:Endocr. J., 2018;65:359-371
9
小野薬品工業:メチロシンのチロシン水酸化酵素阻害(社内資料;2019年1月8日承認、CTD2.6.2.2)
10
小野薬品工業:メチロシンのin vivoカテコールアミン含量減少作用(社内資料;2019年1月8日承認、CTD2.6.2.2)
11
小野薬品工業:メチロシンの循環器に対する作用(社内資料;2019年1月8日承認、CTD2.6.2.2)

