


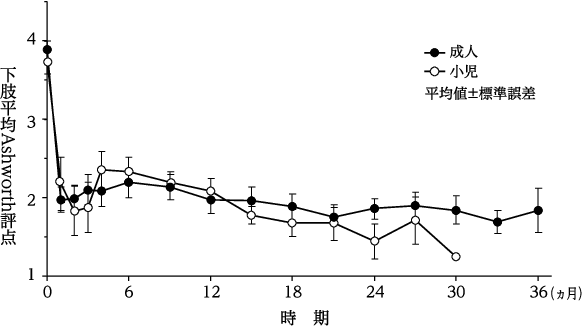
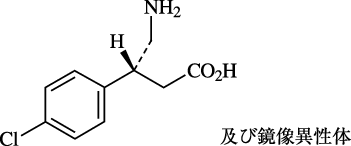
1
社内資料:国内第Ⅲ相試験(DL4040-01試験)(2005年4月11日承認、CTD2.7.2.2)
2
社内資料:薬物動態試験(2005年4月11日承認、CTD2.6.4.4)
3
Faigle JW, et al.:Postgrad Med J. 1972;48(S-5):9-13
4
高杉紀雄ほか:日本薬学会講演要旨集(第97回)1977:237
5
社内資料:薬物動態試験(2005年4月11日承認、CTD2.6.4.6)
6
社内資料:国内臨床成績(2007年1月26日承認、CTD2.7.6)
7
社内資料:海外臨床成績(2005年4月11日承認、CTD2.7.6.5)
8
社内資料:海外臨床成績(2005年4月11日承認、CTD2.7.6.4)
9
Gilmartin R, et al.:J Child Neurol. 2000;15(2):71-77
10
社内資料:海外臨床成績(2005年4月11日承認、CTD2.7.6.7)
11
Meythaler JM, et al.:Arch Phys Med Rehabil. 1996;77(5):461-466
12
山口広貴ほか:臨床医薬 2016;32(6):419-467
13
Schwarz M, et al.:Local-spinal therapy of spasticity. Berlin:Springer-Verlag;1988:65-79
14
Kroin JS, et al.:Exp Brain Res. 1984;54(1):191-194
15
福田英臣ほか:応用薬理 1977;13(5):611-626
16
Fehr HU, et al.:J Int Med Res. 1974;2:36-47
17
Kuroiwa M, et al.:Pharmacol Res. 2009;60(5):392-396
18
津山直一ほか:薬理と治療 1976;4(4):959-970
19
糸賀叡子ほか:診断と治療 1976;64(9):1772-1776
20
玄番央恵ほか:臨床脳波1977;19(6):395-399
21
Wilson PR, et al.:Eur J Pharmacol. 1978;51(4):323-330
22
Yaksh TL, et al.:Anesthesiology 1981;54(6):451-467

