



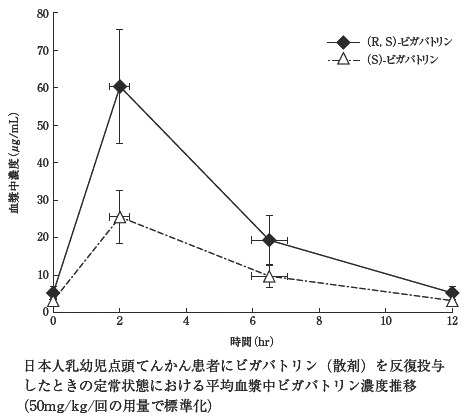
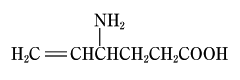
1
Horton M, et al.:J Child Neurol. 2009;24(5):1543-6
2
社内資料:ラットの胚・胎児発生に関する試験(2016年3月28日承認、CTD2.6.6.6)
3
社内資料:ウサギの胚・胎児発生に関する試験(2016年3月28日承認、CTD2.6.6.6)
4
社内資料:ラットの出生前及び出生後の発生並びに母体の機能に関する試験(2016年3月28日承認、CTD2.6.6.6)
5
Williams A, et al.:Aust N Z J Med. 1994;24(1):65
6
社内資料:マウスがん原性試験(2016年3月28日承認、CTD2.6.6.5)
7
社内資料:ラット1年間経口投与毒性試験(回復試験含む)(2016年3月28日承認、CTD2.6.6.3)
8
社内資料:イヌ1年間経口投与毒性試験(2016年3月28日承認、CTD2.6.6.3)
9
社内資料:サル6年間経口投与毒性試験(2016年3月28日承認、CTD2.6.6.3)
10
社内資料:イヌ1年間経口投与毒性試験の回復試験(2016年3月28日承認、CTD2.6.6.3)
11
Walzer M, et al.:Neurotoxicology. 2011;32(6):963-74
12
Rasmussen A D, et al.:Neurotoxicology. 2015;46:137-44
13
Bottomley A L, et al.:Toxicol Pathol. 2015;43(7):1015-24
14
社内資料:ラットの眼毒性試験(2016年3月28日承認、CTD2.6.6.8)
15
社内資料:アルビノラットと有色ラットの網膜への影響の比較(2016年3月28日承認、CTD2.6.6.8)
16
社内資料:有色ラットによる眼毒性試験(2016年3月28日承認、CTD2.6.6.8)
17
Izumi Y, et al.:Epilepsia. 2004;45(9):1043-8
18
社内資料:点頭てんかんを対象とした第3相試験(2016年3月28日承認、CTD2.7.6.2)
19
社内資料:日本人健康成人被験者におけるビガバトリン単回及び反復投与時の安全性及び薬物動態(2016年3月28日承認、CTD2.7.2.2)
20
社内資料:In vitroにおけるビガバトリンの血漿タンパク結合(2016年3月28日承認、CTD2.7.2.2)
21
社内資料:健康被験者に14C-ビガバトリンを単回経口投与したときの薬物動態及び代謝(2016年3月28日承認、CTD2.7.2.2)
22
社内資料:In vitroにおけるビガバトリンの酵素誘導(CYP2B6,2C9,2C19,3A4)(2016年3月28日承認、CTD2.7.2.2)
23
社内資料:In vitroにおけるビガバトリンの酵素誘導(CYP1A2,3A4/5)(2016年3月28日承認、CTD2.7.2.2)
24
社内資料:外国人腎機能障害患者における薬物動態(2016年3月28日承認、CTD2.7.2.2)
25
社内資料:点頭てんかんを対象とした長期投与試験(2016年3月28日承認、CTD2.7.6.2)
26
社内資料:海外第III相単盲検試験(2016年3月28日承認、CTD2.7.6.2)
27
社内資料:海外第III相プラセボ対照二重盲検試験(2016年3月28日承認、CTD2.7.6.2)
28
社内資料:海外第III相クロスオーバー比較試験(他剤比較)(2016年3月28日承認、CTD2.7.6.2)
29
社内資料:In vitroにおけるビガバトリンのGABA-T阻害作用(2016年3月28日承認、CTD2.6.2.2)
30
Jung M J, et al.:J Neurochem. 1977;29(5):797-802
31
Bohlen P, et al.:Brain Res. 1979;167(2):297-305
32
Iadarola M J, et al.:Brain Res Bull. 1980;5:13-9
33
Kubova H, et al.:Epilepsia. 2010;51(3):469-72
34
社内資料:マウスにおける薬物誘発痙攣に対するビガバトリン単回経口投与の作用(2016年3月28日承認、CTD2.6.2.2)
35
社内資料:ビガバトリンの抗痙攣作用(2016年3月28日承認、CTD2.6.2.2)
36
Shin C, et al.:Brain Res. 1986;398(2):370-4
37
Schechter P J, et al.:Eur J Pharmacol. 1977;45(5):319-28

