


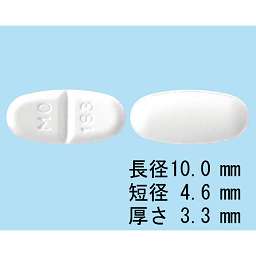
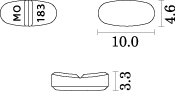
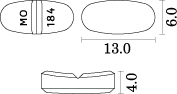
1
Chambers, C. D. et al.:N. Engl. J. Med. 2006;354(6):579-587
2
Källén, B. et al.:Pharmacoepidemiol. Drug Saf. 2008;17(8):801-806
3
Wagner, K. D. et al.:J. Am. Acad. Child Adolesc. Psychiatry. 2006;45(3):280-288
4
持田製薬社内資料:国内第I相試験-エスシタロプラムの単回及び反復投与試験-(2011年4月22日承認、CTD 2.7.6.4.1)
5
持田製薬社内資料:海外臨床薬物動態試験-エスシタロプラムの薬物動態に及ぼす食事の影響-(2011年4月22日承認、CTD 2.7.6.3.2)
6
持田製薬社内資料:海外臨床薬物動態試験-シタロプラムの生物学的利用率-(2011年4月22日承認、CTD 2.7.6.2.1)
7
持田製薬社内資料:薬物動態試験-エスシタロプラムの蛋白結合の検討-(2011年4月22日承認、CTD 2.6.4.4.3、2.6.5.6.2)
8
持田製薬社内資料:薬物動態試験-エスシタロプラムのin vitro代謝の検討-(2011年4月22日承認、CTD 2.6.4.5.2)
9
Olesen, O. V. et al.:Pharmacology. 1999;59(6):298-309
10
Rochat, B. et al.:Biochem. Pharmacol. 1998;56(1):15-23
11
持田製薬社内資料:海外臨床薬物動態試験-シタロプラムのマスバランス-(2011年4月22日承認、CTD 2.7.6.4.5)
12
持田製薬社内資料:海外臨床薬物動態試験-腎機能障害患者におけるシタロプラムの薬物動態-(2011年4月22日承認、CTD 2.7.6.5.5)
13
持田製薬社内資料:海外臨床薬物動態試験-肝機能障害患者におけるエスシタロプラムの薬物動態-(2011年4月22日承認、CTD 2.7.6.5.4)
14
持田製薬社内資料:海外臨床薬物動態試験-高齢者におけるエスシタロプラムの薬物動態(単回投与)-(2011年4月22日承認、CTD 2.7.6.5.1)
15
持田製薬社内資料:海外臨床薬物動態試験-高齢者におけるエスシタロプラムの薬物動態(反復投与)-(2011年4月22日承認、CTD 2.7.6.5.2)
16
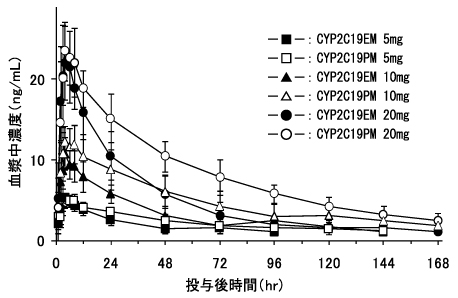
持田製薬社内資料:海外臨床薬物動態試験-エスシタロプラムの薬物動態に及ぼすCYP2D6遺伝子多型の影響-(2011年4月22日承認、CTD 2.7.2.3)
17
持田製薬社内資料:海外臨床薬物動態試験-エスシタロプラム及びシタロプラムの薬物相互作用試験-(2011年4月22日承認、CTD 2.7.6.6)
18
持田製薬社内資料:用量反応非劣性試験-大うつ病性障害患者におけるプラセボ及び塩酸パロキセチンを対照とした有効性及び安全性の検討-(2011年4月22日承認、CTD 2.7.6.8.2)
19
持田製薬社内資料:長期投与試験-大うつ病性障害患者における長期投与の安全性及び有効性の検討-(2011年4月22日承認、CTD 2.7.6.9.1)
20
持田製薬社内資料:高齢者長期投与試験-大うつ病性障害患者における長期投与の安全性、有効性及び薬物動態の検討-(2011年4月22日承認、CTD 2.7.6.9.2)
21
持田製薬社内資料:社会不安障害に対するプラセボ対照試験(2015年11月20日承認、CTD 2.7.6.1.1)
22
持田製薬社内資料:社会不安障害に対する長期投与試験(2015年11月20日承認、CTD 2.7.6.2.1)
23
持田製薬社内資料:海外Thorough QT試験-エスシタロプラムの心臓再分極に及ぼす影響-(2011年4月22日承認、CTD 2.7.6.7.4)
24
持田製薬社内資料:薬理試験-うつ病モデル及び不安障害モデルにおける有効性-(2011年4月22日承認、CTD 2.6.2.2.1、2.6.2.3.1)
25
Sánchez, C. et al.:Psychopharmacology. 2003;167(4):353-362
26
Montgomery, S. A. et al.:Pharmacol. Toxicol. 2001;88(5):282-286
27
Sánchez, C. et al.:Behav. Pharmacol. 2003;14(5-6):465-470
28
持田製薬社内資料:薬理試験-ラット社会的ストレスモデルの行動様式に及ぼす影響-(2011年4月22日承認、CTD 2.6.2.2.1)
29
持田製薬社内資料:薬理試験-ラット脳シナプトソームの5-HT取り込み(in vitro)及びテトラベナジン誘発によるマウスの行動(in vivo)に及ぼす影響-(2011年4月22日承認、CTD 2.6.2.2.2、2.6.2.3.2)
30
Mørk, A. et al.:Neuropharmacology. 2003;45(2):167-173
31
Owens, M. J. et al.:Biol. Psychiatry. 2001;50(5):345-350
32
Hyttel, J. et al.:J. Neural Transm. Gen. Sect. 1992;88(2):157-160
33
持田製薬社内資料:薬理試験-エスシタロプラム及び代謝物のモノアミン取り込みに及ぼす影響(in vitro及びin vivo)-(2011年4月22日承認、CTD 2.6.2.2.2、2.6.2.3.2)
34
持田製薬社内資料:薬理試験-各種受容体及びトランスポータに対するリガンドの結合に及ぼす影響-(2011年4月22日承認、CTD 2.6.2.2.2、2.6.2.3.2)

