


1
FACT SHEET FOR HEALTH CARE PROVIDERS EMERGENCY USE AUTHORIZATION(EUA)OF REMDESIVIR(GS-5734TM)
2
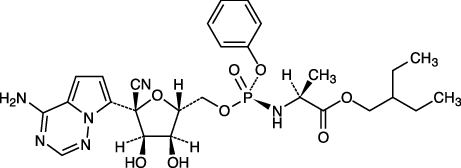
社内資料(レムデシビル治験薬概要書)
3
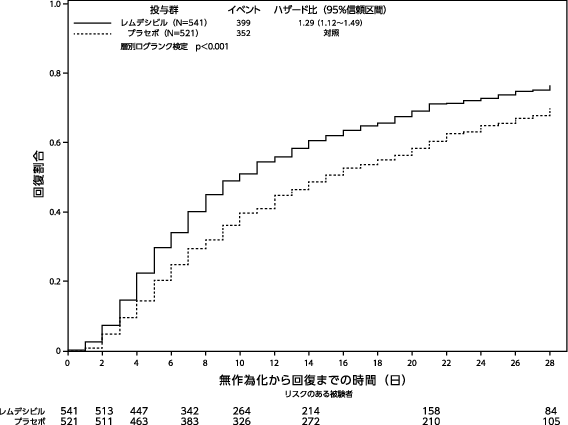
社内資料(NIAID ACTT-1試験)
4
社内資料(GS-US-540-5773試験)
5
Goldman JD, et al. N Engl J Med. 2020;383(19):1827-1837
6
社内資料(GS-US-540-5774試験)
7
Spinner CD, et al. JAMA 2020;324(11):1048-1057
8
Pruijssers AJ, et al. Cell Rep. 2020;32(3):107940
9
Xie X, et al. Nat Commun. 2020;11(1):5214
10
社内資料(PC-540-2026試験、PC-540-2034試験、PC-540-2038試験、PC-540-2039試験)
11
社内資料(PC-540-2028試験)
12
社内資料(PC-540-2029試験)
13
Gottlieb RL, et al. N Engl J Med. Published online December 22, 2021. doi:10.1056/NEJMoa2116846
14
社内資料(GS-US-540-9013試験)
15
社内資料(REP-22251 In-Use安定性試験)
16
社内資料(PC-540-2033試験)
17
社内資料(PC-540-2040試験)
18
社内資料(GS-US-540-5823試験)

