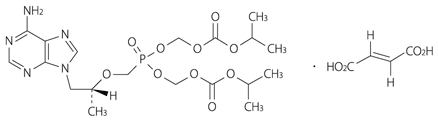
1
Benaboud S,et al.:Antimicrob Agents Chemother.2011;55:1315-1317
2
中道昇:新薬と臨牀.2005;54(8):941-948
3
Perry CM,et al.:Drugs.2009;69:2245-2256

抗ウイルス化学療法剤
1錠 644.4円
有効成分 | 1錠中 テノホビル ジソプロキシルフマル酸塩 300mg |
|---|---|
添加剤 | 部分アルファー化デンプン、クロスカルメロースナトリウム、乳糖水和物、結晶セルロース、ステアリン酸マグネシウム、ヒプロメロース、酸化チタン、トリアセチン |
| 剤形・性状 | 白色のフィルムコーティング錠 |
|---|---|
| 識別コード | GSK 300 |
| 表 (長径×短径) | 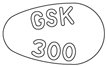 17.0mm×10.5mm |
| 裏 |  |
| 側面 (厚さ) |  5.3mm |
| 質量 | 693mg |
クレアチニンクリアランス | 投与方法 |
50mL/min以上 | 300mgを1日1回 |
30~49mL/min | 300mgを2日に1回 |
10~29mL/min | 300mgを3~4日に1回 |
血液透析患者 | 300mgを7日に1回注)又は累積約12時間の透析終了後に300mgを投与 |
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
ジダノシン | 膵炎、乳酸アシドーシス等のジダノシンによる副作用を増強するおそれがあるので、ジダノシンの減量を考慮すること。 | 機序不明だが、ジダノシンのAUC及びCmaxが上昇する。 |
アタザナビル硫酸塩 | アタザナビルの治療効果が減弱するおそれがあるので、本剤とアタザナビル硫酸塩を併用する場合には、本剤とアタザナビル300mgをリトナビル100mgとともに投与することが望ましい。また、本剤による副作用を増強するおそれがある。 | 機序不明だが、アタザナビルのAUC、Cmax及びCminが低下し、テノホビルのAUC、Cmax及びCminが上昇する。 |
ロピナビル・リトナビル | 本剤による副作用を増強するおそれがある。 | 機序不明だが、テノホビルのAUC及びCminが上昇する。 |
アシクロビル バラシクロビル塩酸塩 ガンシクロビル バルガンシクロビル塩酸塩 | これらの薬剤又は本剤による副作用を増強するおそれがある。 | 尿細管への能動輸送により排泄される薬剤と併用する場合、排泄経路の競合により、排泄が遅延し、これらの薬剤又は本剤の血中濃度が上昇するおそれがある。 |
レジパスビル・ソホスブビル | 本剤とレジパスビル・ソホスブビルとの併用により、テノホビルの血漿中濃度が上昇する。 | 作用機序は不明であるが、本剤が基質となるPgp及びBCRPに対するレジパスビルの阻害作用が関与すると考えられる。 |
ベルパタスビル・ソホスブビル | 本剤とベルパタスビル・ソホスブビルとの併用により、テノホビルの血漿中濃度が上昇する。 | 作用機序は不明であるが、本剤が基質となるPgp及びBCRPに対するベルパタスビルの阻害作用が関与すると考えられる。 |
腎毒性を有する薬剤 | 併用は避けることが望ましい。 | 腎毒性を有する薬剤は腎機能障害の危険因子となる。 |
1~5%未満 | 1%未満 | 頻度不明 | |
|---|---|---|---|
消化器 | 悪心、腹痛 | 下痢、嘔吐、鼓腸 | |
腎臓 | 蛋白尿、多尿 | ||
肝臓 | 肝炎 | ||
過敏症 | アレルギー反応(血管浮腫) | ||
代謝 | 低カリウム血症、低リン酸血症、体脂肪の再分布/蓄積 | ||
筋骨格 | 骨軟化症(骨痛、骨折)、ミオパチー | ||
臨床検査 | 肝機能検査値異常(AST、ALT及びγ-GTP増加等)、クレアチニン増加、アミラーゼ増加、リパーゼ増加 | ||
その他 | 発疹 | 浮動性めまい、呼吸困難、無力症 |
CLcr (mL/min) | 例数 | Cmax (ng/mL) | AUC(0-inf) (ng・hr/mL) | CL/F (mL/min) | CLr (mL/min) |
>80 | 3 | 335.5±31.8 | 2184.5 ±257.4 | 1043.7±115.4 | 243.5±33.3 |
50~80 | 10 | 330.4±61.0 | 3063.8 ±927.0 | 807.7±279.2 | 168.6±27.5 |
30~49 | 8 | 372.1±156.1 | 6008.5 ±2504.7 | 444.4±209.8 | 100.6±27.5 |
<30 (12~28)注1) | 11 | 601.6±185.3 | 15984.7 ±7223.0 | 177.0±97.1 | 43.0±31.2 |
末期腎不全 患者 (透析前) | 9 | 1061±252.8 | 44900.8 ±12956.8注2) | - | - |
末期腎不全 患者 (透析後) | 8 | 904.5±326.3 | 15768.1 ±5366.3注2) | - | - |
LOC115409(109例) | ||
評価時点 | 24週時 | 48週時 |
投与前HBe抗原 | 陽性及び陰性 | |
投与前平均HBV-DNA値±標準偏差(log10 copies/mL) | 7.00±1.498(109例) | |
HBV-DNAの投与前値からの平均変化量±標準偏差 (log10 copies/mL) | -4.57±1.122 (109例) | -4.86±1.353 (109例) |
HBV-DNA陰性化率注1) | 54%(59/109) | 77%(84/109) |
ALT正常化率注2) | 70%(58/83) | 75%(62/83) |
セロコンバージョン率注3) | 0%(0/43) | 9%(4/43) |
GS-US-174-0102注4) | ||
評価時点 | 48週時(250例) | 240週時(375例) |
投与前HBe抗原 | 陰性 | |
投与前平均HBV-DNA値±標準偏差(log10 copies/mL) | 6.86±1.308(250例) | |
HBV-DNAの投与前値からの平均変化量±標準偏差(log10 copies/mL) | -4.57±1.347 (241例) | -4.65±1.294 (295例) |
HBV-DNA陰性化率注1) | 91.2%(228/250) | 98.6%(291/295) |
ALT正常化率注2) | 76.3%(180/236) | 85.2%(236/277) |
セロコンバージョン率注3) | - | - |
GS-US-174-0103注4) | ||
評価時点 | 48週時(176例) | 240週時(266例) |
投与前HBe抗原 | 陽性 | |
投与前平均HBV-DNA値±標準偏差(log10 copies/mL) | 8.64±1.076(176例) | |
HBV-DNAの投与前値からの平均変化量±標準偏差(log10 copies/mL) | -6.17±1.067 (160例) | -6.30±1.141 (165例) |
HBV-DNA陰性化率注1) | 68.8%(121/176) | 96.6%(169/175) |
ALT正常化率注2) | 68.0%(115/169) | 73.4%(124/169) |
セロコンバージョン率注3) | 20.9%(32/153) | 40.2%(66/164) |
LOC115912(34例) | ||
評価時点 | 24週時 | 48週時 |
投与前HBe抗原 | 陽性及び陰性 | |
投与前平均HBV-DNA値±標準偏差(log10 copies/mL) | 5.57±1.739(34例) | |
HBV-DNAの投与前値からの平均変化量±標準偏差 (log10 copies/mL) | -3.09±1.432 (34例) | -3.26±1.586 (34例) |
HBV-DNA陰性化率注1) | 59%(20/34) | 62%(21/34) |
ALT正常化率注2) | 60%(9/15) | 53%(8/15) |
セロコンバージョン率注3) | 0%(0/28) | 0%(0/28) |
GS-US-174-0106(53例) | ||
評価時点 | 48週時 | 168週時 |
投与前HBe抗原 | 陽性及び陰性 | |
投与前平均HBV-DNA値±標準偏差(log10 copies/mL) | 6.06±1.430(53例) | |
HBV-DNAの投与前値からの平均変化量±標準偏差(log10 copies/mL) | -3.58±1.290 (52例) | -3.79±1.305 (44例) |
HBV-DNA陰性化率注1) | 75.5%(40/53) | 80.4%(41/51) |
ALT正常化率注2) | 40.7%(11/27) | 68.0%(17/25) |
セロコンバージョン率注3) | 5.3%(2/38) | 13.5%(5/37) |
GS-US-174-0121(141例) | |
評価時点 | 96週時 |
投与前HBe抗原 | 陽性及び陰性 |
投与前平均HBV-DNA値±標準偏差(log10 copies/mL) | 6.40±1.826(141例) |
HBV-DNAの投与前値からの平均変化量±標準偏差(log10 copies/mL) | -4.16±1.785(132例) |
HBV-DNA陰性化率注1) | 85.8%(121/141) |
ALT正常化率注2) | 62.0%(49/79) |
セロコンバージョン率注3) | 10.8%(7/65) |