

 200mg(カプセル)、
200mg(カプセル)、
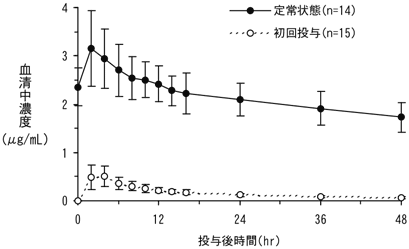
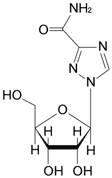
1
深瀬広幸ほか. 臨床医薬. 2002;18:521-38.
2
社内資料:反復投与(2004年10月22日承認、CTD 2.7.2.2)
3
社内資料:食事の影響(2001年11月21日承認、申請資料概要ヘ.4.(5))
4
社内資料:バイオアベイラビリティ(2001年11月21日承認、申請資料概要ヘ.4.(4))
5
社内資料:肝機能障害患者(2001年11月21日承認、申請資料概要ヘ.4.(8))
6
社内資料:腎機能障害患者(2001年11月21日承認、申請資料概要ヘ.4.(9))
7
社内資料:血漿蛋白結合率(2001年11月21日承認、申請資料概要ヘ.3.(2).3))
8
社内資料:ヒトにおける体内動態(2001年11月21日承認、申請資料概要ヘ.4.(1))
9
社内資料:組織内分布(2001年11月21日承認、申請資料概要ヘ.3.(2).1))
10
社内資料:胎盤・胎児移行性(2001年11月21日承認、申請資料概要ヘ.3.(2).2))
11
社内資料:代謝:ラット(2001年11月21日承認、申請資料概要ヘ.3.(3).1).①)
12
社内資料:代謝:サル(2001年11月21日承認、申請資料概要ヘ.3.(3).1).②)
13
社内資料:チトクロムP450に対する阻害作用(2001年11月21日承認、申請資料概要ヘ.3.(3).2).①)
14
社内資料:胆汁中排泄(2001年11月21日承認、申請資料概要ヘ.3.(4).2))
15
社内資料:乳汁移行性(2001年11月21日承認、申請資料概要ヘ.3.(4). 3))
16
社内資料:肝薬物代謝酵素誘導(2001年11月21日承認、申請資料概要ヘ.3.(3).2).②)
17
Glue P, et al. Hepatology. 2000;32:647-53.
18
社内資料:制酸剤の影響(2001年11月21日承認、申請資料概要ヘ.4.(6))
19
社内資料:レプリコンを用いたリバビリンの評価試験
20
社内資料:リバビリンの抗HCV作用機序(2001年11月21日承認、申請資料概要ホ.1.(2).7))
21
社内資料:抗ウイルス作用を裏付ける試験(2004年10月22日承認、CTD 2.6.2.2)

