


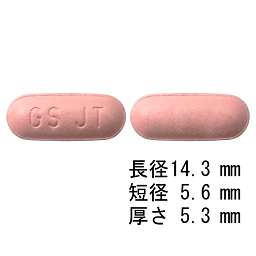



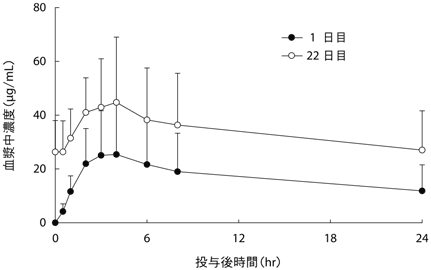
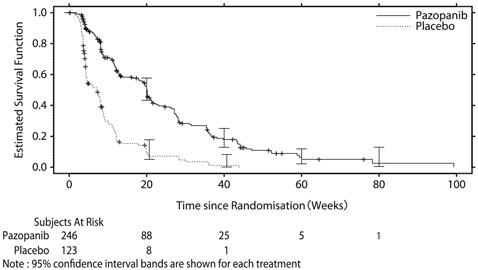
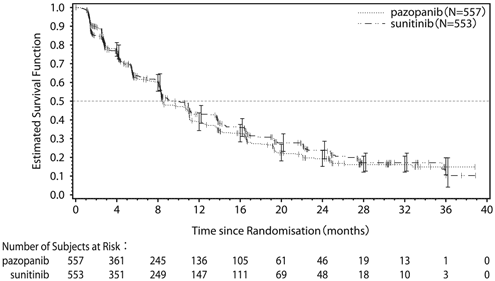
1
NDBを用いた調査結果の概要(VEGF/VEGFR阻害作用を有する薬剤の動脈解離に関するリスク評価):https://www.pmda.go.jp/files/000266521.pdf
2
Inada-Inoue, M. et al.:Cancer Chemother. Pharmacol. 2014;73:673-683[20153683]
3
社内資料:海外第Ⅰ相試験(VEG10004) (2012年9月28日承認、CTD2.7.6 VEG10004試験)[20155743]
4
社内資料:海外第Ⅰ相試験(VEG10005) (2012年9月28日承認、CTD2.7.6 VEG10005試験)[20155740]
5
社内資料:分布に関する試験(1) (2012年9月28日承認、CTD2.7.2.2.1.1)[20155741]
6
社内資料:分布に関する試験(2) (2012年9月28日承認、CTD2.7.2.2.1.2)[20155742]
7
社内資料:分布に関する試験(3) (2012年9月28日承認、CTD2.6.4.4.6)[20155744]
8
社内資料:分布に関する試験(4) (2012年9月28日承認、CTD2.6.4.4.7)[20155745]
9
社内資料:代謝に関する試験(1) (2012年9月28日承認、CTD2.7.2.2.1.4)[20155746]
10
社内資料:海外第Ⅰ,Ⅱ及びⅢ相試験(母集団薬物動態) (2012年9月28日承認、CTD2.7.2.3.2)[20155832]
11
社内資料:海外第Ⅰ相試験(NCI-8063) (2012年9月28日承認、CTD2.7.6 NCI-8063試験)[20155747]
12
社内資料:相互作用に関する試験(1) (2012年9月28日承認、CTD 2.6.4.5.5.1.1)[20155748]
13
社内資料:海外第Ⅰ相試験(VEG10007) (2012年9月28日承認、CTD2.7.6 VEG10007試験)[20155749]
14
社内資料:相互作用に関する試験(2) (2012年9月28日承認、CTD2.6.4.4.10)[20155750]
15
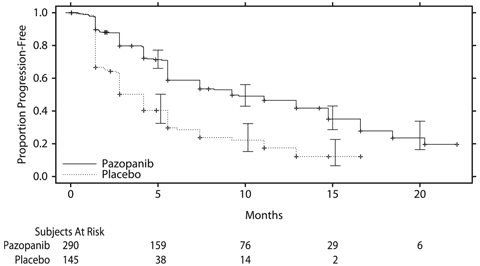
社内資料:国際共同第Ⅲ相試験(VEG110727) (2012年9月28日承認、CTD2.7.6 VEG110727試験)[20155751]
16
社内資料:海外第Ⅱ相試験(VEG20002) (2012年9月28日承認、CTD2.7.6 VEG20002試験)[20155752]
17
van Glabbeke, M. et al.:Eur. J. Cancer. 2002;38:543-549 [20152755]
18
社内資料:国際共同第Ⅲ相試験(VEG108844) (2014年3月17日承認、CTD2.7.6 VEG108844試験)[20155753]
19
社内資料:海外第Ⅲ相試験(VEG105192) (2014年3月17日承認、CTD2.7.6 VEG105192試験)[20155754]
20
Kumar, R. et al.:Mol. Cancer Ther. 2007;6:2012-2021 [20153573]
21
Hosaka, S. et al.:J. Orthop. Res. 2012;30:1493-1498 [20153574]
22
社内資料:薬効薬理試験(1) (2012年9月28日承認、CTD 2.6.2.2.2.3.1)[20155755]
23
社内資料:薬効薬理試験(2) (2012年9月28日承認、CTD 2.6.2.2.2.3.2)[20155756]

