


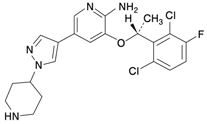
1
社内資料:胚・胎児発生に関する試験 (2012年3月30日承認、CTD2.6.6.6)[L20110830222]
2
社内資料:チトクロームP450に対する阻害作用 (2012年3月30日承認、CTD2.6.4.7)[L20110830213]
3
社内資料:排出トランスポーターに対する阻害作用 (2012年3月30日承認、CTD2.6.4.7)[L20120308001]
4
社内資料:有機イオントランスポーターを介した薬物相互作用のin vitro評価[L20131212153]
5
社内資料:ミダゾラムとの薬物相互作用 (2012年3月30日承認、CTD2.7.2.2)[L20120308002]
6
社内資料:イトラコナゾールとの薬物相互作用[L20180316001]
7
社内資料:リファンピシンとの薬物相互作用 (2012年3月30日承認、CTD2.7.2.2)[L20110830245]
8
社内資料:ラットにおける反復投与毒性試験 (2012年3月30日承認、CTD2.6.6.3)[L20120308003]
9
社内資料:遺伝毒性試験 (2012年3月30日承認、CTD2.6.6.4)[L20120308004]
10
社内資料:日本人健康成人における薬物動態(単回投与) (2012年3月30日承認、CTD2.7.2.2)[L20110830242]
11
社内資料:日本人癌患者における薬物動態(単回投与・反復投与) (2012年3月30日承認、CTD5.3.5.2)[L20110830227]
12
社内資料:バイオアベイラビリティ (2012年3月30日承認、CTD2.7.1.2)[L20110830239]
13
社内資料:食事の影響 (2012年3月30日承認、CTD2.7.1.2)[L20110830241]
14
社内資料:分布に関与する非臨床パラメータ (2012年3月30日承認、CTD2.6.4.4)[L20110830210]
15
社内資料:代謝酵素 (2012年3月30日承認、CTD2.6.4.5)[L20110830211]
16
社内資料:代謝・排泄経路 (2012年3月30日承認、CTD2.7.2.2)[L20110830243]
17
社内資料:腎機能障害を伴う被験者における薬物動態 [L20131212154]
18
社内資料:肝機能障害を伴う被験者における薬物動態 [L20170803004]
19
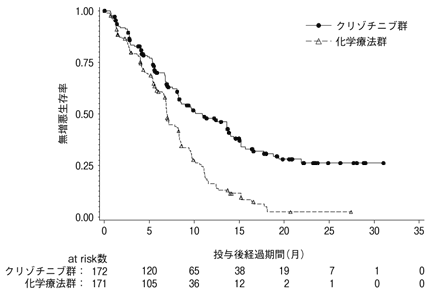
Solomon BJ, et al.:N Engl J Med.2014;371(23):2167-2177
20
社内資料:国際共同第Ⅲ相試験(A8081007試験)[L20131212155]
21
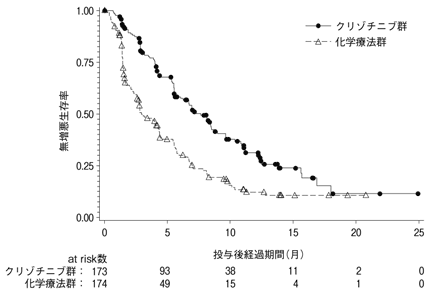
Shaw AT, et al.:N Engl J Med.2013;368(25):2385-2394
22
社内資料:国際共同第Ⅱ相試験(A8081005試験) (2012年3月30日承認、CTD2.7.3.3、2.7.4.6)[L20110830248]
23
社内資料:海外第Ⅰ相試験(A8081001試験) (2012年3月30日承認、CTD2.7.3.2、2.7.4.2)[L20110830247]
24
社内資料:国際共同第Ⅱ相試験(OO12-01試験) (2017年5月18日承認、CTD2.7.6.1)[L20170301214]
25
社内資料:非臨床薬理試験(in vivo) (2012年3月30日承認、CTD2.6.2.2)[L20110830203]
26
社内資料:非臨床薬理試験(in vitro) (2012年3月30日承認、CTD2.6.2.2)[L20120308005]

