










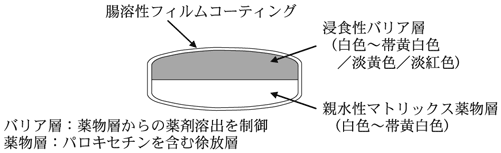
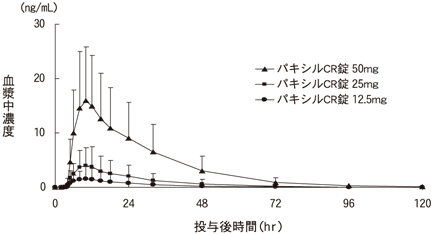
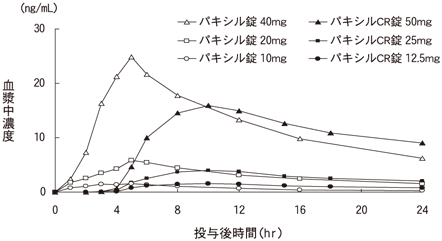
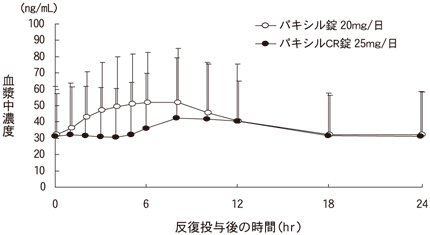
1
Chambers CD,et al.:N Engl J Med.2006;354:579-587
2
Kallen B,et al.:Pharmacoepidemiol Drug Saf.2008;17:801-806
3
Öhman R,et al.:J Clin Psychiatry.1999;60:519-523
4
入江 廣ほか:薬理と治療.2000;28(Suppl 1):47-68
5
Crewe HK,et al.:Br J Clin Pharmacol.1992;34:262-265
6
Özdemir V,et al.:Clin Pharmacol Ther.1997;62:334-347
7
Albers LJ,et al.:Psychiatry Res.1996;59:189-196
8
Hemeryck A,et al.:Clin Pharmacol Ther.2000;67:283-291
9
Sindrup SH,et al.:Clin Pharmacol Ther.1992;51:278-287
10
Kaye CM,et al.:Acta Psychiatr Scand Suppl.1989;350:60-75
11
Dalhoff K,et al.:Eur J Clin Pharmacol.1991;41:351-354
12
永田良一ほか:薬理と治療.2000;28(Suppl 1):89-110
13
Higuchi T,et al.:Psychiatry Clin Neurosci.2011;65:655-663
14
Thomas DR,et al.:Psychopharmacology.1987;93:193-200
15
Gartside SE,et al.:Br J Pharmacol.1995;115:1064-1070
16
Lassen JB:Psychopharmacology.1978;57:151-153
17
Kennett GA,et al.:Neuropharmacology.1994;33:1581-1588
18
Perrault GH,et al.:Pharmacol Biochem Behav.1992;42:45-47
19
島田 瞭ほか:実中研・前臨床研究報.1996;20:163-167
20
Lightowler S,et al.:Pharmacol Biochem Behav.1994;49:281-285
21
Cadogan AK,et al.:Br J Pharmacol.1992;107(Proc Suppl Oct):108P

