


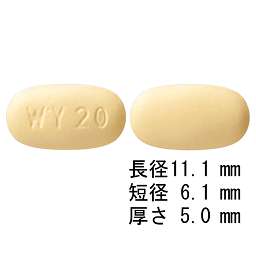


1
社内資料:臨床薬理試験(個々の試験結果の要約)(2010年7月23日承認、CTD2.7.2.2)[L20100506093]
2
社内資料:閉経後骨粗鬆症患者における薬物動態(2010年7月23日承認、CTD2.7.3.2.1.2)[L20100630065]
3
社内資料:生物薬剤学及び関連する分析法(個々の試験結果の要約)(2010年7月23日承認、CTD2.7.1.2)[L20100506091]
4
社内資料:分布(2010年7月23日承認、CTD2.6.4.4)[L20100506078]
5
社内資料:代謝(2010年7月23日承認、CTD2.6.4.5)[L20100506079]
6
社内資料:臨床薬理試験(試験を通した結果の比較及び分析)(2010年7月23日承認、CTD2.7.2.3)[L20100506094]
7
Itabashi A. et al.:J Bone Miner Res. 2011;26 (3):519-529[L20110217004]
8
Silverman S L. et al.: J Bone Miner Res. 2008;23 (12):1923-1934[L20100406200]
9
社内資料:薬理試験(効力を裏付ける試験)(2010年7月23日承認、CTD2.6.2.2)[L20100506073]

