1
社内資料:血漿中濃度(2005年7月25日承認、申請資料概要へ2.1.1)[L70010000054]
2
社内資料:代謝と排泄(2005年7月25日承認、申請資料概要へ2.2.2.4)[L70010000055]
3
社内資料:副作用集計[L70010000056]
4
Hamann Philip R. et al.:Bioconjug Chem.2002;13(1):47-58
5
Naito K. et al.:Leukemia.2000;14(8):1436-1443

抗悪性腫瘍剤抗腫瘍性抗生物質結合抗CD33モノクローナル抗体
1瓶 202239円
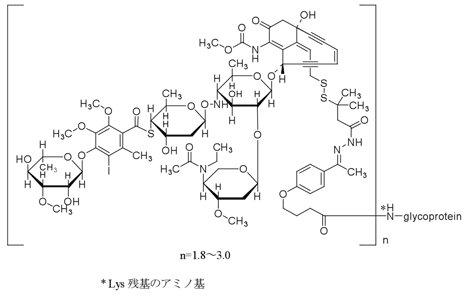
有効成分 | 1バイアル中 ゲムツズマブオゾガマイシン(遺伝子組換え) 5mg |
|---|---|
添加剤 | 精製白糖 77.7mg デキストラン40 45.5mg 塩化ナトリウム 29.2mg リン酸二水素ナトリウム一水和物 0.5mg 無水リン酸一水素ナトリウム 3.0mg |
| pH | 7.0~7.6[1mg/mL注射用水] |
|---|---|
| 浸透圧比 (生理食塩液に対する比) | 約0.8 [1mg/mL注射用水] |
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
副腎皮質ホルモン メチルプレドニゾロン等 マクロライド系抗生物質 ジョサマイシンプロピオン酸エステル等 ケトライド系抗生物質 テリスロマイシン ストレプトグラミン系抗生物質 キヌプリスチン・ダルホプリスチン 抗真菌剤 イトラコナゾール等 | 臨床症状については不明である。 | 本剤はCYP3A4により代謝される可能性が示唆されているため、これらの薬剤が本剤の代謝に影響を及ぼす可能性がある。 |
10%以上 | 5〜10%未満 | 5%未満 | |
|---|---|---|---|
皮膚 | 発疹 | そう痒、毛包炎、爪囲炎 | |
消化器 | 悪心(59.3%)、嘔吐(48.9%)、食欲不振、下痢、腹痛、便秘 | 歯肉出血 | 消化不良、歯周炎、メレナ、腹部膨満、吐血、口渇、胃炎、口唇炎、しゃっくり、血腫(口唇、口腔内) |
精神・神経 | めまい | 不眠、しびれ(感覚鈍麻)、不安、抑うつ、浮遊感(異常感) | |
呼吸器 | 咳嗽、咽頭炎 | ラ音、鼻炎、嗄声、呼吸音の変化、喉頭炎 | |
循環器 | 不整脈(頻脈等)、低血圧 | 高血圧 | 心拍数減少、動悸、心電図異常、心不全、心筋虚血 |
血液 | 点状出血、凝固線溶系異常 | 斑状出血、紫斑、皮下出血 | |
代謝異常 | LDH上昇、低カリウム血症 | 低カルシウム血症、低リン酸血症、低アルブミン血症、高血糖 | 低蛋白血症、尿酸減少、低ナトリウム血症、低マグネシウム血症、尿酸増加、高トリグリセリド血症、高カリウム血症、高コレステロール血症、低コレステロール血症、低血糖、BUN減少、高カルシウム血症、低クロール血症、低トリグリセリド血症、高クロール血症、高ナトリウム血症、高リン酸塩血症 |
生殖器 | 腟出血、不正子宮出血 | ||
その他 | 発熱(77.0%)、悪寒(60.6%)、頭痛、脱力感、倦怠感 | 浮腫、体重減少、疼痛(耳痛、四肢痛、肛門周囲痛)、筋痛、胸痛、投与部位反応(炎症、感染、出血)、背部痛、体重増加、味覚異常、関節痛、ほてり、顔面腫脹、冷感 |
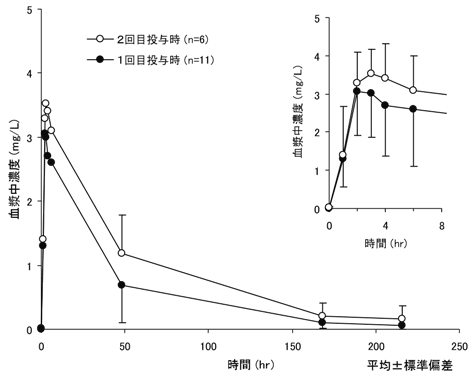
投与回数 | Cmax (mg/L) | tmax (hr) | AUC0〜∞ (mg・hr/L) | t1/2 (hr) |
1回目 | 3.248±1.195 | 2.02(中央値) | 133.4±94.0 | 51±25 |
2回目 | 3.640±0.859 | 3.02(中央値) | 223.1±135.9 | 59±36 |
試験番号 | 201 | 202 | 203 | 合計 |
N | 84 | 95 | 98 | 277 |
完全寛解症例数 (%) | 14 (17%) | 13 (14%) | 8 (8%) | 35 (13%) |
95%信頼区間 | 9-26% | 7-22% | 4-15% | 9-17% |