



1
医療上の必要性の高い未承認薬・適応外薬検討会議 公知申請への該当性に係る報告書:カペシタビン(進行性胃癌)
2
医療上の必要性の高い未承認薬・適応外薬検討会議 公知申請への該当性に係る報告書:カペシタビン(直腸癌における補助化学療法)
3
Cavaliere A, et al. Tumori. 1990;76:179-81.
4
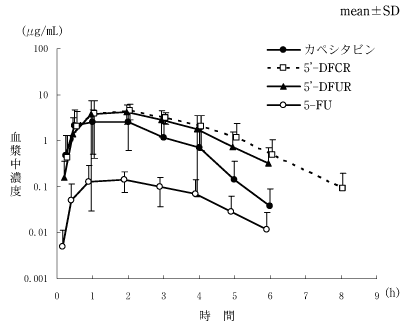
日本人患者における薬物動態(国内前期臨床第2相試験)(2003年4月16日承認、申請資料概要ヘ.3-2)
5
日本人患者における薬物動態(国内臨床第1相試験)(2003年4月16日承認、申請資料概要ヘ.3-1)
6
Hyodo I, et al. Jpn J Clin Oncol. 2006;36:410-7.
7
臓器、組織中濃度(2003年4月16日承認、申請資料概要ヘ.2-2-1)
8
胎児移行性(2003年4月16日承認、申請資料概要ヘ.2-2-3)
9
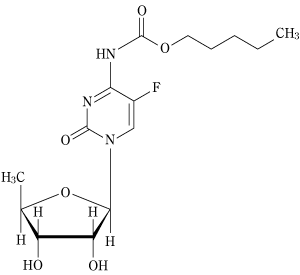
カペシタビンの5-FUへの腫瘍選択的変換(2003年4月16日承認、申請資料概要ホ-1.3-2)
10
代謝経路(2003年4月16日承認、申請資料概要ヘ.2-3-1)
11
Judson IR, et al. Invest New Drugs. 1999;17:49-56.
12
腎機能障害を伴う固形癌患者の薬物動態(2003年4月16日承認、申請資料概要ヘ.3-3-7)
13
Camidge R, et al. J Clin Oncol. 2005;23:4719-25.
14
肝ミクロソーム薬物代謝酵素系に対する影響(2003年4月16日承認、申請資料概要ヘ.2-3-4)
15
Reigner B, et al. Cancer Chemother Pharmacol. 1999;43:309-15.
16
進行・再発乳癌を対象とした前期第2相臨床試験(2003年4月16日承認、申請資料概要ト.1-2-1)
17
進行・再発乳癌を対象とした後期第2相臨床試験(2003年4月16日承認、申請資料概要ト.1-3-1)
18
ドセタキセル無効の進行・再発乳癌を対象とした後期第2相臨床試験(2003年4月16日承認、申請資料概要ト.1-3-2)
19
社内資料:タキサン系薬剤無効の乳癌を対象とした第2相臨床試験
20
Blum JL, et al. J Clin Oncol. 1999;17:485-93.
21
社内資料:パクリタキセル又はドセタキセル無効の進行・再発乳癌を対象とした第2相臨床試験
22
Twelves C, et al. N Engl J Med. 2005;352:2696-704.
23
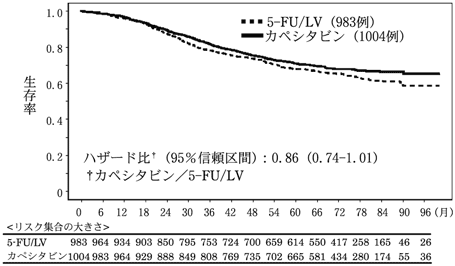
社内資料:Dukes Cの結腸癌を対象とした術後補助化学療法の第3相臨床試験(単剤投与)
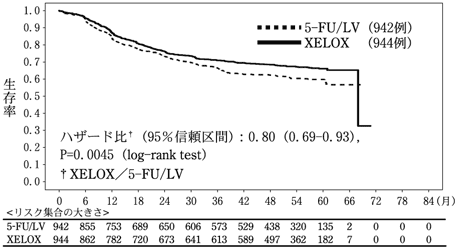
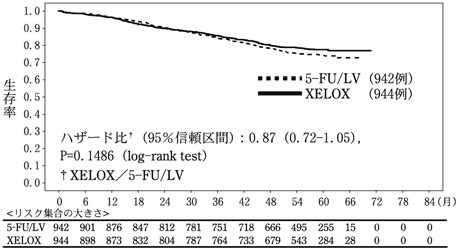
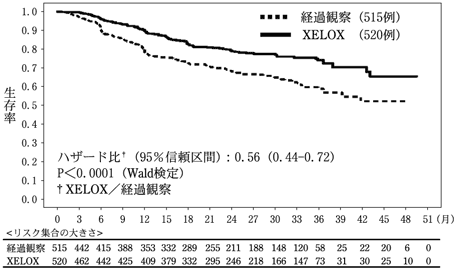
24
社内資料:Dukes Cの結腸癌を対象とした術後補助化学療法の第3相臨床試験(併用投与)
25
社内資料:進行・転移性結腸・直腸癌を対象とした第1/2相臨床試験
26
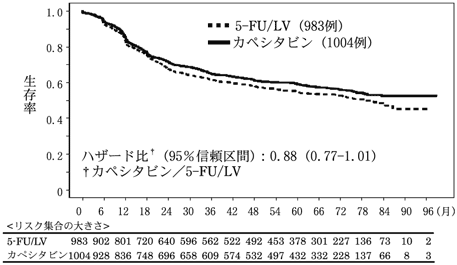
社内資料:転移性結腸・直腸癌を対象とした第3相臨床試験
27
社内資料:イリノテカン・フルオロウラシル・ホリナート療法の治療歴がある転移性結腸・直腸癌を対象とした第3相臨床試験
28
Bang YJ, et al. Lancet. 2012;379:315-21.
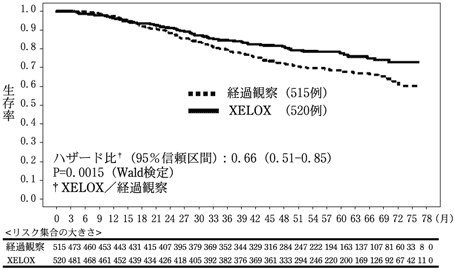
29
Noh SH, et al. Lancet Oncol. 2014;15:1389-96.
30
作用部位・作用機序(2003年4月16日承認、申請資料概要ホ-1.1-1)
31
Pinedo HM, et al. J Clin Oncol. 1988;6:1653-64.
32
抗腫瘍効果(2003年4月16日承認、申請資料概要ホ-1.4-1)
33
Yanagisawa M, et al. Oncol Rep. 2009;22:241-7.
34
Sawada N, et al. Oncol Rep. 2007;18:775-8.

