

1
社内資料:日本人患者における薬物動態(2018年9月21日承認、CTD 2.7.2.3)
2
社内資料:腎機能障害患者における薬物動態(2018年9月21日承認、CTD 2.7.2.3)
3
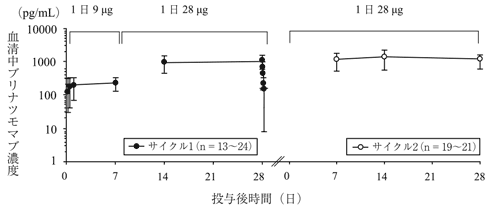
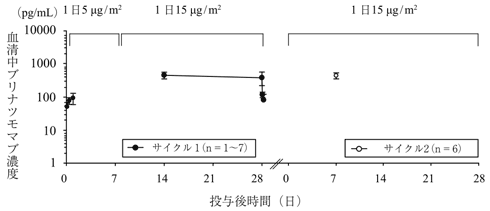
社内資料:20130265試験 日本人第Ib/Ⅱ相試験(2018年9月21日承認、CTD 2.7.6.11)
4
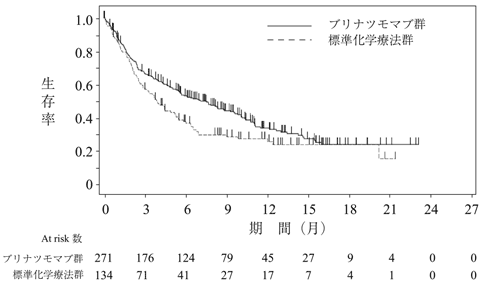
社内資料:00103311試験 海外第Ⅲ相試験(2018年9月21日承認、CTD 2.7.6.1)
5
Kantarjian H, et al.:N Engl J Med. 2017;376:836-847
6
社内資料:MT103-205試験 海外第I/Ⅱ相試験(2018年9月21日承認、CTD 2.7.6.5)
7
von Stackelberg A, et al.:J Clin Oncol. 2016;34:4381-4389
8
社内資料:20120216試験 海外第Ⅱ相試験(2018年9月21日承認、CTD 2.7.6.4)
9
Martinelli G, et al.:J Clin Oncol. 2017;35:1795-1802
10
Dreier T, et al.:Int J Cancer. 2002;100:690-697
11
社内資料:103-PCD-0076試験 白血病由来細胞株を用いたin vitroでの検討(2018年9月21日承認、CTD 2.6.2.3)
12
社内資料:103-PCD-0099試験 腫瘍細胞株移植モデルを用いた検討(2018年9月21日承認、CTD 2.6.2.3)
13
Dreier T, et al.:J Immunol. 2003;170:4397-4402

