

1
Ogura M, et al. Cancer Sci. 2013;104:105-10.
2
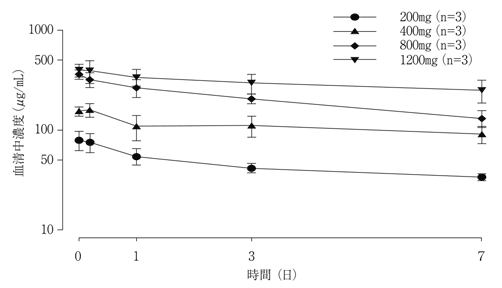
海外第Ⅰb相試験(BO21000試験)(2018年7月2日承認、申請資料概要2.7.2.2.6)
3
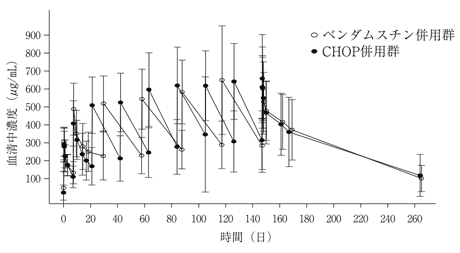
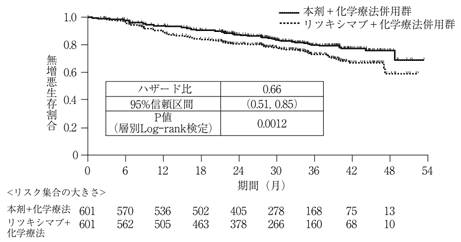
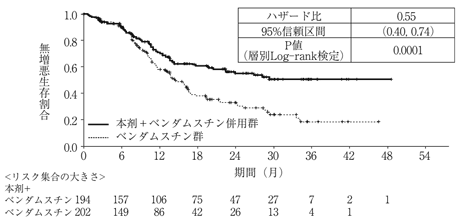
国際共同第Ⅲ相比較試験(BO21223試験)(2018年7月2日承認、申請資料概要2.7.3.2.1)
4
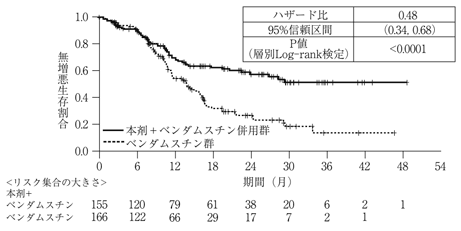
海外第Ⅲ相比較試験(GAO4753g試験)(2018年7月2日承認、申請資料概要2.7.3.2.2)
5
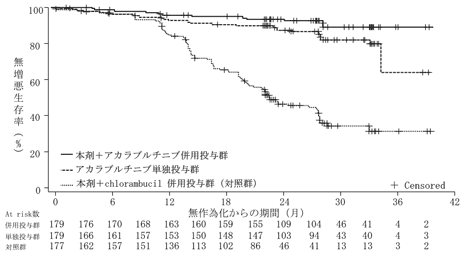
社内資料:国際共同第Ⅳ相試験(MO40597試験)
6
Sharman JP, et al. Lancet. 2020; 395(10232): 1278-1291
7
Izutsu K, et al. Cancer Sci. 2021; 112(6):2405-2415
8
社内資料:国内第Ⅰ相試験(D8220C00001試験)
9
Mössner E, et al. Blood. 2010;115:4393-402.
10
Herter S, et al. Mol Cancer Ther. 2013;12:2031-42.
11
マクロファージ/単球によるADCC/ADCP活性(2018年7月2日承認、申請資料概要2.6.2.2.2.3(3))

