

1
医療上の必要性の高い未承認薬・適応外薬検討会議 公知申請への該当性に係る報告書:リツキシマブ(遺伝子組換え)(免疫抑制状態下のCD20陽性のB細胞性リンパ増殖性疾患(成人))
2
医療上の必要性の高い未承認薬・適応外薬検討会議 公知申請への該当性に係る報告書:リツキシマブ(遺伝子組換え)(免疫抑制状態下のCD20陽性のB細胞性リンパ増殖性疾患(小児))
3
Krysko KM, et al.:Neurol. Neuroimmunol. Neuroinflamm. 2020;7(1):e637
4
Igarashi T,et al.:Ann. Oncol. 2002;13:928-943
5
Igarashi T,et al.:Int.J.Hematol. 2001;73:213-221
6
Tobinai K,et al.:Ann. Oncol. 1998;9:527-534
7
Alasfoor K, et al.:Ann. Hematol. 2009;88:239-243
8
「関節リウマチ患者(国内未承認)を対象とした外国第Ⅱ相試験(GP13-201試験)」(社内資料)
9
「CD20陽性のB細胞性非ホジキンリンパ腫におけるIDEC-C2B8薬物動態」(リツキサン®点滴静注 添付文書)
10
「IDEC-C2B8単回投与時の薬物動態と腫瘍移行性」(リツキサン®点滴静注、承認年月日:2001年6月20日、申請資料概要ヘ. 吸収、分布、代謝、排泄)
11
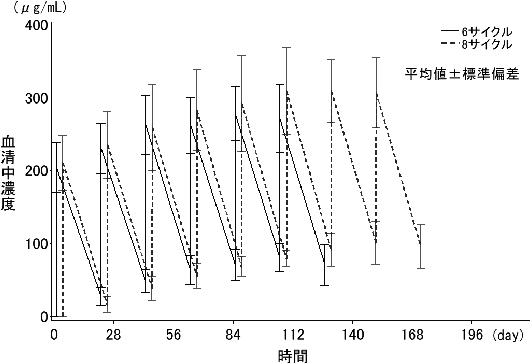
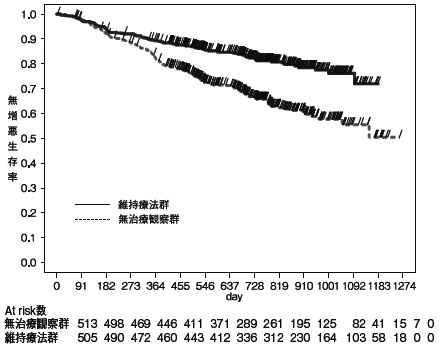
「進行期ろ胞性リンパ腫患者を対象とした国際共同第Ⅲ相試験(GP13-301試験)」(社内資料)
12
「IDEC-C2B8 CD20陽性のB細胞性非ホジキンリンパ腫国内臨床試験及び国外臨床試験の概要」(リツキサン®点滴静注 添付文書)
13
「IDEC-C2B8 未治療CD20陽性のろ胞性非ホジキンリンパ腫に対する維持療法の海外臨床試験の概要」(リツキサン®点滴静注 添付文書)
14
「IDEC-C2B8 再発又は難治性のろ胞性非ホジキンリンパ腫に対する他の抗悪性腫瘍剤との併用の海外臨床試験の概要」(リツキサン®点滴静注 添付文書)
15
Stone J. et al.:N. Engl. J. Med. 2010;363:221-232
16
「In vitro 生物機能の品質試験」(社内資料)
17
「全血を用いたB細胞枯渇の比較評価(GP13-021試験)」(社内資料)
18
「In vitro ADCC効力の比較評価(GP13-017試験)」(社内資料)
19
「In vitro ADCC効力の比較評価(GP13-022試験)」(社内資料)
20
「FcγRⅡ/RⅢ介在性ADCCの評価(GP13-PBMC試験)」(社内資料)
21
「組織交差反応性アッセイ(GP13-010試験)」(社内資料)
22
「単回静脈内投与PK/PD比較試験(GP13-007試験)」(社内資料)
23
「反復投与毒性比較試験(GP13-008試験)」(社内資料)
24
「マウス異種移植腫瘍モデルを用いた抗腫瘍効果比較試験(GP13-011試験)」(社内資料)
25
「マウス異種移植腫瘍モデルを用いた抗腫瘍効果及びPDの比較試験(GP13-014試験)」(社内資料)
26
「マウス異種移植腫瘍モデルを用いた生存比較試験(GP13-015試験)」(社内資料)
27
「マウス異種移植腫瘍モデルを用いた生存比較試験(GP13-018試験)」(社内資料)
28
Reff ME,et al.:Blood.1994;83:435-445
29
「マウス抗体IDEC-2B8のヒトCD20に対する結合特異性」(リツキサン®点滴静注、承認年月日:2001年6月20日、申請資料概要 ホ. 薬理作用)
30
「マウス-ヒトキメラ抗体IDEC-C2B8のヒト末梢血中Bリンパ球及びヒト原発性Bリンパ腫細胞に対する結合特異性」(リツキサン®点滴静注、承認年月日:2001年6月20日、申請資料概要 ホ. 薬理作用)
31
「ヒト正常組織との交叉反応性試験」(リツキサン®点滴静注、承認年月日:2001年6月20日、申請資料概要ニ. 毒性)

